There's (quite rightly) a lot of raving about the new AlphaFold DB resource today. Thought I'd weigh in with my own example. DNA-dependent protein kinase catalytic subunit (uniprot.org/uniprot/P78527) is an enormous beast - a single chain of 4,128 residues! (1/10)
https://twitter.com/PDBeurope/status/1418511467989684224
There are now 19 structures of this guy in the wwPDB, all post-resolution-revolution cryoEM structures. The first (2017) was reconstructed to a character-building 4.3 Å. I'm told the model building was extremely challenging, ... (2/10)
... and a glance at the overall structure shows why. The thing is almost entirely composed of short helix after short helix after short helix... without clear sidechain information getting out of step with the map would be really, really easy. (3/10) 

Anyway, about 6 months ago I happened to be idly looking at one of the more recent higher-resolution (mid-3 Å) structures in ISOLDE, and picked out (and corrected) a few out-of-register helices. I spotted a few other issues and meant to go through the whole thing, ... (4/10)
... but never found the few days of time I expected it would take. Anyway, the AlphaFold DB announcement brought it back to mind. To my initial disappointment P78527 didn't come up in a search, but then I read the fine print: (5/10)
"... we have attempted to fold most sequences in the UniProt reference proteome that are between 16 and 2700 amino acids long and contain only standard amino acids.For human proteins only, longer sequences are available split into fragments in the bulk download." (6/10)
So I downloaded the full database, and sure enough there are no less than 14 overlapping models of 1400 residues each, covering the entire chain (cryo-EM model in CA trace, AlphaFold models as ribbons). (7/10)
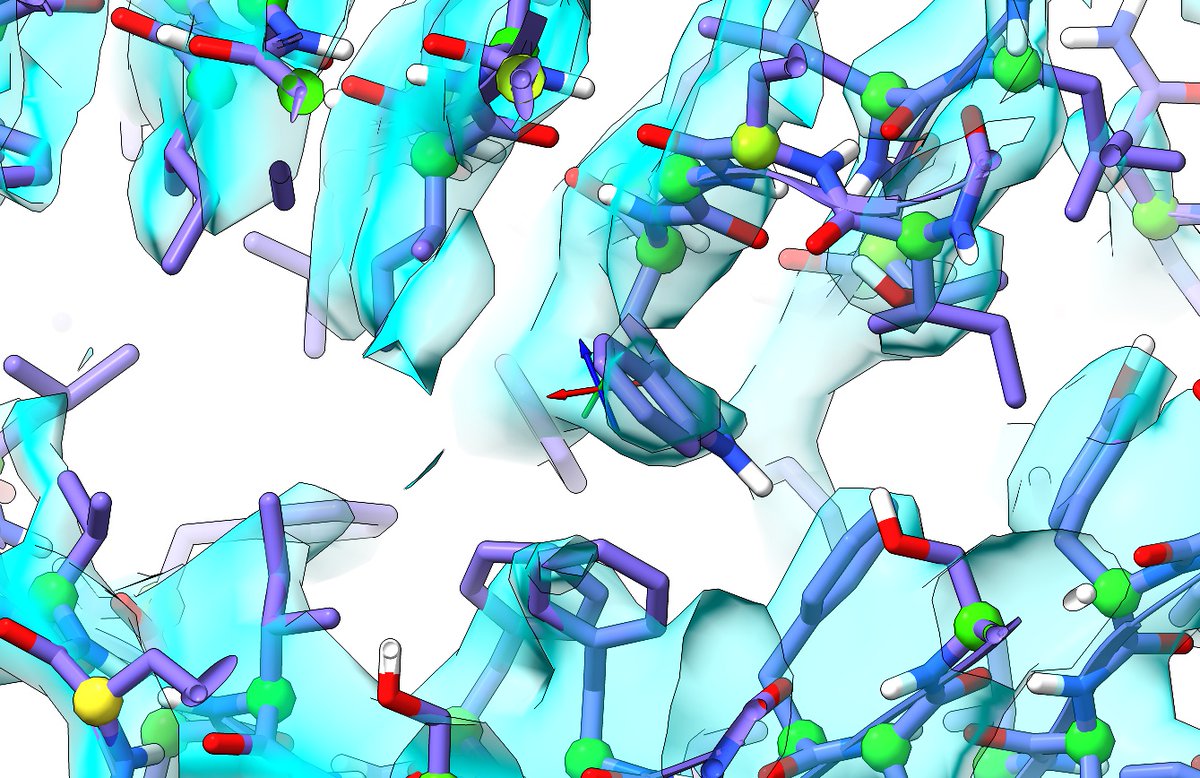
Taking a subset of the AlphaFold models that together provide complete coverage and using these as templates for ISOLDE's adaptive distance and torsion restraints, and rebuilding the cryo-EM model becomes almost trivial. (8/10)
So far, for every site of disagreement between cryo-EM and AlphaFold models (with the exception of long-range interdomain contacts), it's been the AlphaFold one that was closest to correct. For example, this little site here. (9/10)
Upshot: while AlphaFold clearly isn't a *replacement* for experimental structures by any stretch, it's already very clear that it's going to make the task of *building* experimental structures both much easier and much less error prone. Welcome to the future! (fin)
• • •
Missing some Tweet in this thread? You can try to
force a refresh