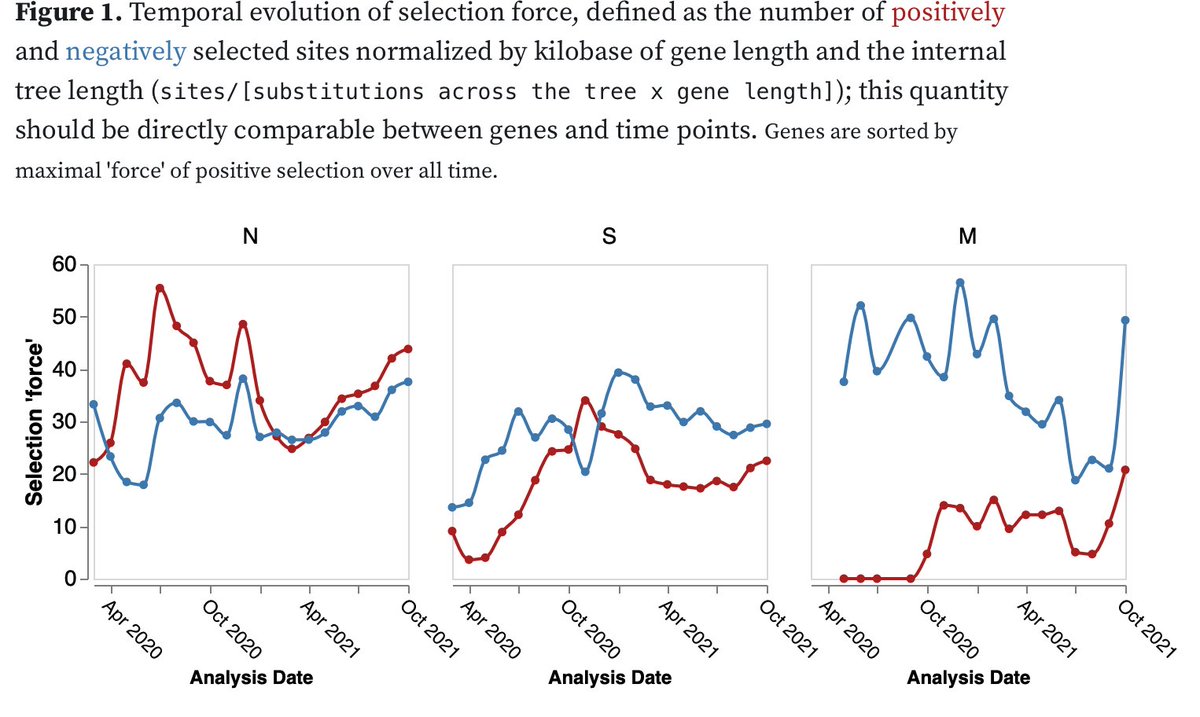
There's definitely a strong signal of selection on Spike in #Omicron compared to reference clades in our preliminary RASCL analysis of ~60 sequences (thanks @aglucaci, more coming)
1. Spike is under positive selection
2. Spike is under stronger selection than background
1. Spike is under positive selection
2. Spike is under stronger selection than background
There are 9 spike sites where there's stronger selection in #Omicron compared to other clades according to Contrast-FEL (academic.oup.com/mbe/article/38…). Sorted by q-value here (stronger evidence at the top) 
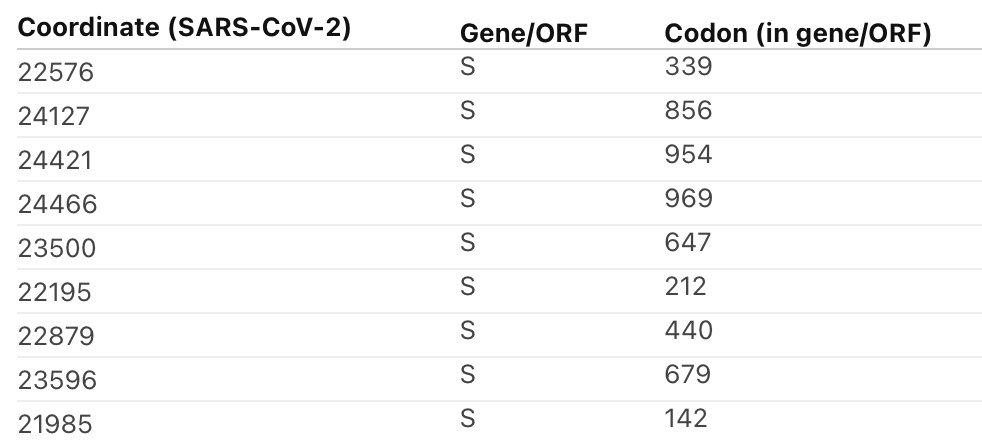
Full details at observablehq.com/@aglucaci/sc2-… Will post further updates (we will be running daily or so updates as more sequences come in).
The selection is all on the basal branch to the clade. Will be very interesting to see how it continues to evolve (assuming the variant spreads). I would expect that some of the mutations (if indeed fixed due to intra-host selection) may revert.
• • •
Missing some Tweet in this thread? You can try to
force a refresh