
Comme promis j'ai lue la partie que je maitrise le mieux du papier.
Donc de la page 4 a 14 des supplementary information (sa fais toujours plaisir d'être reléguer en annexe des papier majeurs :) )
Y a des choses très intéressante :
Donc de la page 4 a 14 des supplementary information (sa fais toujours plaisir d'être reléguer en annexe des papier majeurs :) )
Y a des choses très intéressante :
Notamment l'idée de ne pas corriger les reads mais de les patchés.
Avec les reads de troisièmes génération on a un gros problème c'est le taux d'erreur important.
Le séquenceur ne vas pas lire les bonnes bases mais y a plusieurs type d'erreur différents.
Avec les reads de troisièmes génération on a un gros problème c'est le taux d'erreur important.
Le séquenceur ne vas pas lire les bonnes bases mais y a plusieurs type d'erreur différents.
Les erreurs d'une seul base on vas lire un C a la place d'un T par exemple.
Mais aussi des erreurs structurel:
- des "sequencing jump" on vas sauté une partie de la séquence.
- des chimères, on vas fusionné deux reads, donc relié deux régions du gènome qui n'ont aucun liens
Mais aussi des erreurs structurel:
- des "sequencing jump" on vas sauté une partie de la séquence.
- des chimères, on vas fusionné deux reads, donc relié deux régions du gènome qui n'ont aucun liens
Généralement les assembleurs de troisième génération vont commencé par corriger/éliminer ces erreurs avant de faire quoi que ce soit d'autre et évidement sa prend beaucoup de temps car il faut aligné les reads les un contre les autres et généré un consensus.
Et on espère que ce consensus soi proche de la vérité ou d'une vérité, parce que si on on travail sur un génome hétérozygote la correction peut faire disparaitre cette hétérozygotie même si des assembleurs comme FALCON tant de réglé ce problème.
La plus tôt que de corrigé tout les erreurs MARVEL (l'assembleur crée ? utilisé pour cette étude) vas juste détecter corriger les erreurs structurel, ce qui est pratique parce que sa demande beaucoup moins d'overlap.
Ici on a un sequencing jump, A est le reads qu'on souhaite corriger B1 et B2 deux reads qui nous indique le sequencing jump. Le read A vas être coupé et on vas inséré la bonne séquence au millieu 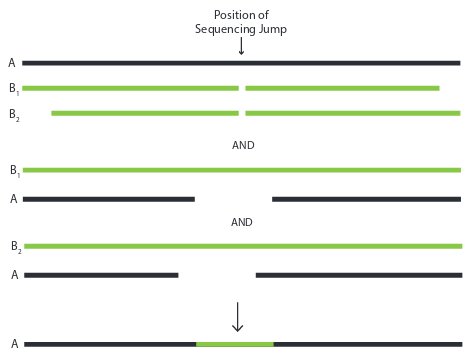
Ici c'est un autre type d'erreur le séquenceur a intégré un peut n'importe quoi mais sa avoir trop confiance en lui, la qualité est faible bar noir au dessus de la séquence. On réalise la même opération que pour le sequencing jump. 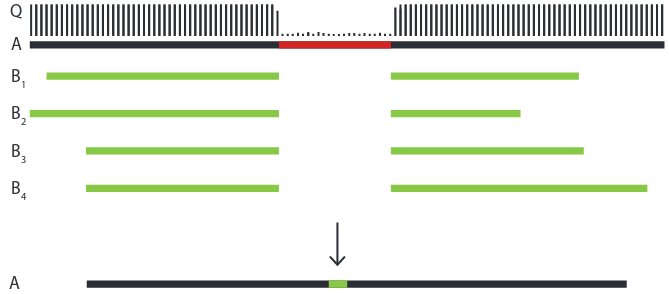
Un dernier cas pour la route. sa resemble beaucoup au cas précédant sauf que le mapping des autres reads n'est pas intérompue. On a sur le read A une région avec une très faible qualité mais les reads B la couvre on peut remplacé la régions de faible qualité. 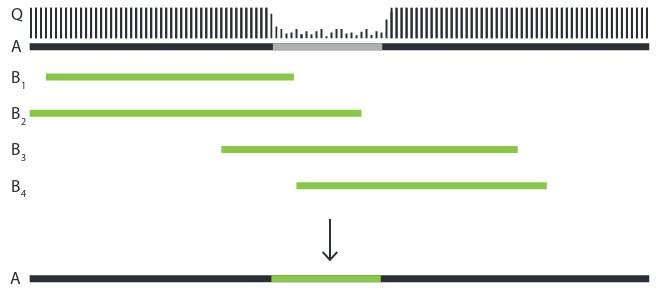
Je vous est mis les figures qu'il y avait dans l'article je vous encourage a le lire, pour plus de détaile nature.com/articles/natur…
En gros le patching des long reads permet d'éliminer d'éliminer les erreurs qui vont gêner l'assemblage et uniquement c'est erreur là du coup il y a un gain de temps en calcule et en mémoire utilisé.
La deuxième gros innovations et le masking dynamique des région répété durant la phase d'overlapping des reads. En gros pour corriger les reads on doit détecter ce qui on une partie commun qui partage la même séquence.
Cette phase d'overlapping elle vas aussi être utile pour l'assemblage on vas reconstruire la séquence d'origine en fussionnant les reads qui partage des partis commune (ou là le gros raccourci pédagogique).
Les régions répété vue que ce sont des duplications on vas avoir beaucoup
Les régions répété vue que ce sont des duplications on vas avoir beaucoup
d'overlap. C'est overlap apporte peut d'information on déjà beaucoup d'autre, donc est-ce que c'est nécessaire d'en rajouté et en plus les régions répété on a souvent du mal a les résoudre durant l'assemblage.
Du coup MARVEL utilise un serveur de repeat masking. En gros avant de voir si un read a des overlaps il vas l'envoyer au serveur le serveur il vas étudier rapidement le reads et si il contient des répétition il vas les masquer.
Après avoir calculer les overlaps du read MARVEL vas les envoyer au server pour qu'il alimente sa base de donnée de régions répété. Plus MARVEL vas avancé dans la détection d'overlap moins il vas perdre de temps sur les régions répété.
En plus ce serveur de régions répété vas être réutilisé durant la construction du graphe d'assemblage pour indiquer les régions ou l’assembleur pourrait commettre des erreurs d'assemblage (relié deux régions du génome qui sont normalement éloigné)
Je suis pas sur d'avoir toujours été très claire ou même super pédagogue désoler. Mais bon sa existe si vous avez des questions hésité pas. De nouveau le liens de l'article nature.com/articles/natur…






