
New preprint from our lab on whole animal multiplexed #scRNAseq (WHAM-seq) by Tara Chari, Brady Weissbourd & @JaseGehring et al. collaborating w/ David Anderson, Evelyn Houliston @Clytia_Vlfr, and @richcopley. A 🧵 about our proof of principle in🎐... 1/ biorxiv.org/content/10.110…
We show #scRNAseq can be used for "reverse genomics" to conduct low-cost *experiments*. Instead of sequence first ask questions later, we ask questions first & then sequence. We illustrate the approach w/ a starvation experiment using the emerging model Clytia hemisphaerica. 2/ 
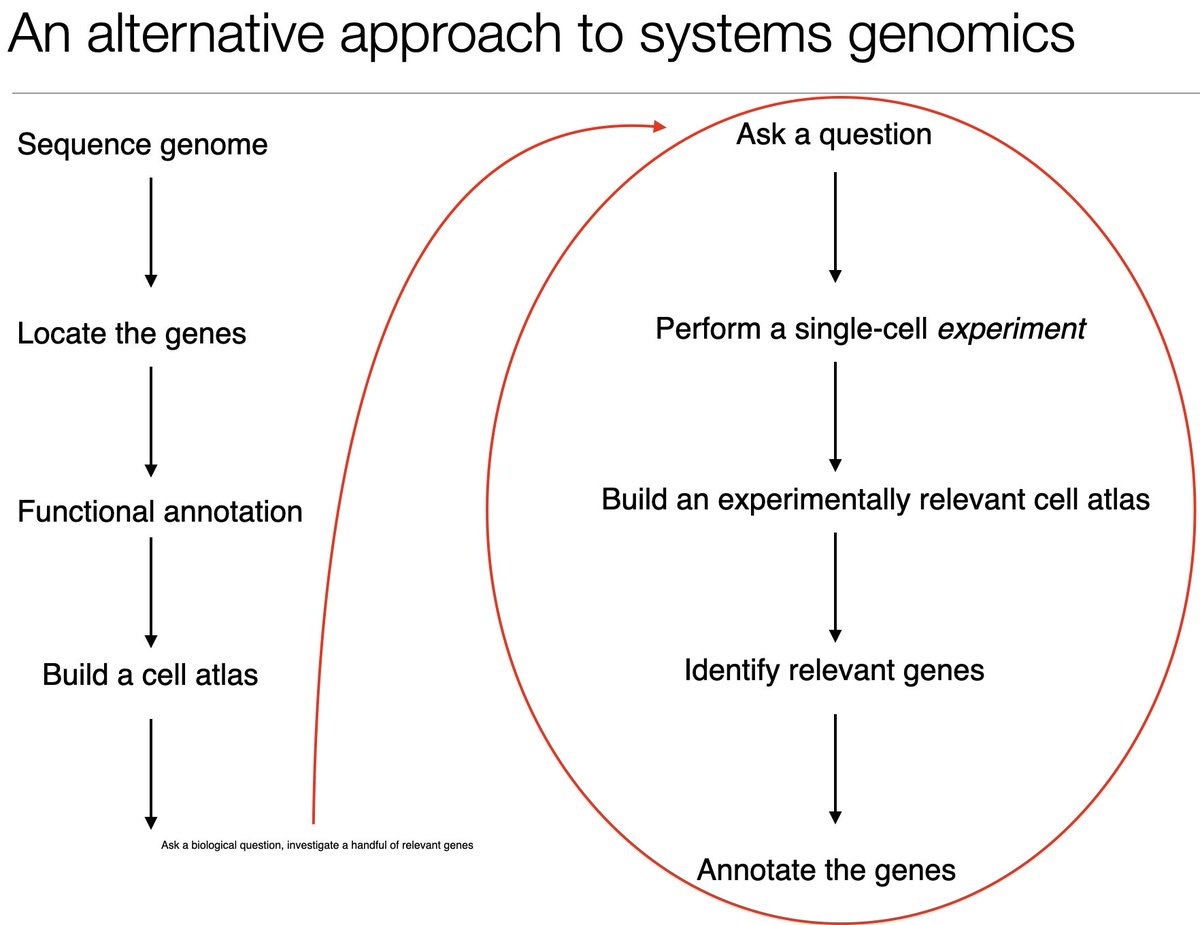
We performed multiplexed #scRNAseq using the ClickTag approach developed in our lab by @JaseGehring (w/@sisichen, Matt Thomson, Jeff Park). The chemical multiplexing can be used on any tissue/animal and facilitates experiments with little batch effect. 3/ nature.com/articles/s4158…
Single-cell RNA-seq had not been perform on Clytia Hemisphaerica prior to our work, so we first had to figure out how to make good cell suspensions. This is one part of #scRNAseq that requires trial and error. @LOLCats4U and @JaseGehring solved it w/ a dounce homogeneizer. 4/

We multiplexed 5 starved and 5 fed jellyfish, and first built a cell atlas. This was both challenging & interesting. E.g., we uncovered a previously unknown putative mechanosensory cell type in Clytia expressing homologs of components characterized in vertebrate hair cells. 5/
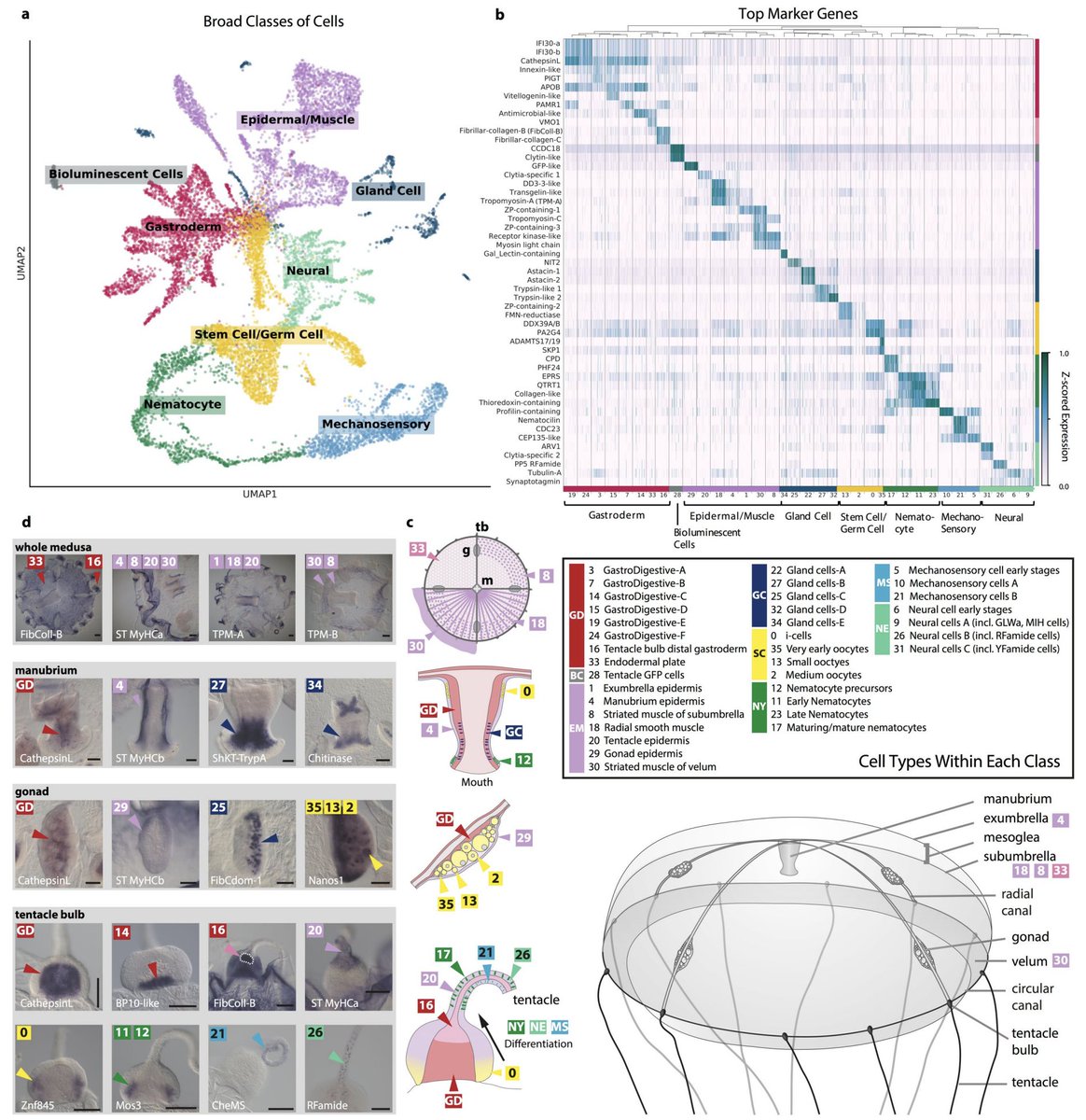
A remarkable feature of the Clytia medusa is that it constantly generates many cell types, notably neural cells and nematocytes from prominent interstitial stem cell pools in the tentacle bulb epidermis. We can see the differentiating cell trajectories in our cell atlas. 6/
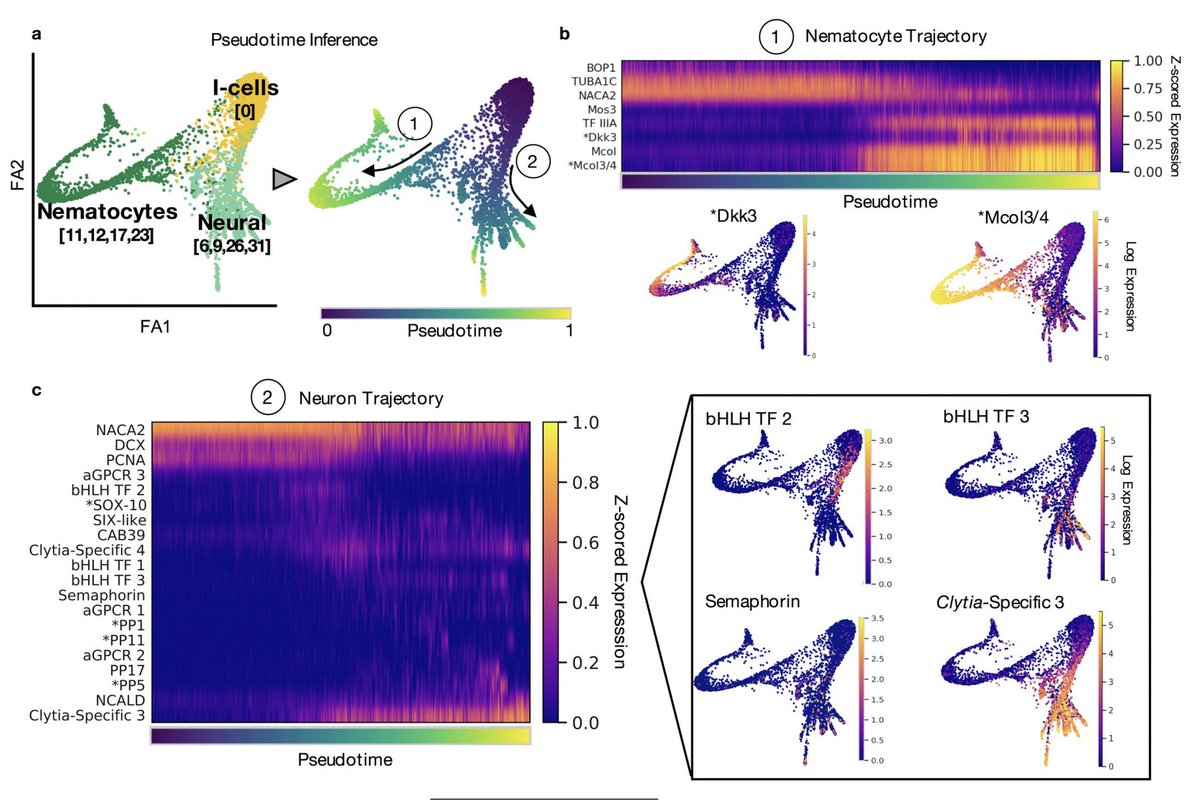
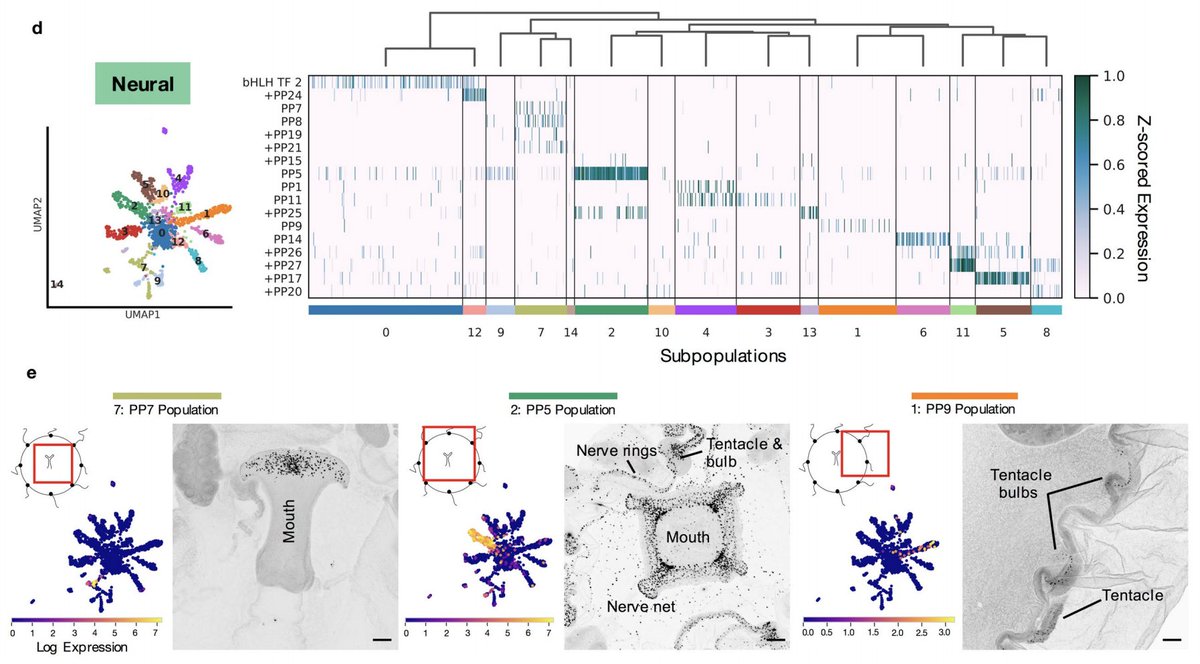
We also identified 14 likely subpopulations of neurons, and 10 new likely neuropeptides. The neuronal subpopulations appear to express combinatorial combinations of neuropeptides, and some are spatially localized whereas others are broadly distributed across the animal. 7/
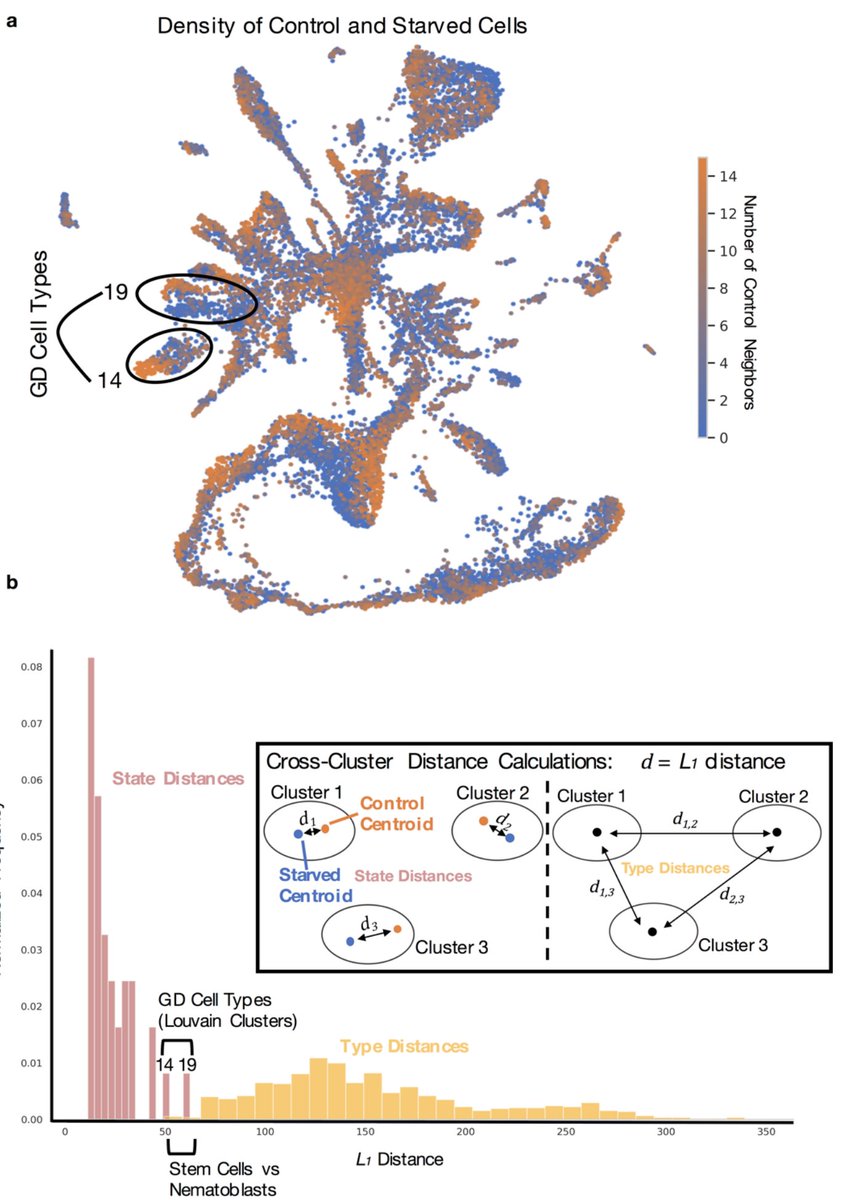
The starvation experiment revealed an interesting story of shifts in cell *state* as opposed to cell *type*. We believe we are sensitive to such shifts because of the greatly reduced batch effect in our experiments as a result of multiplexing our samples with ClickTags. 8/
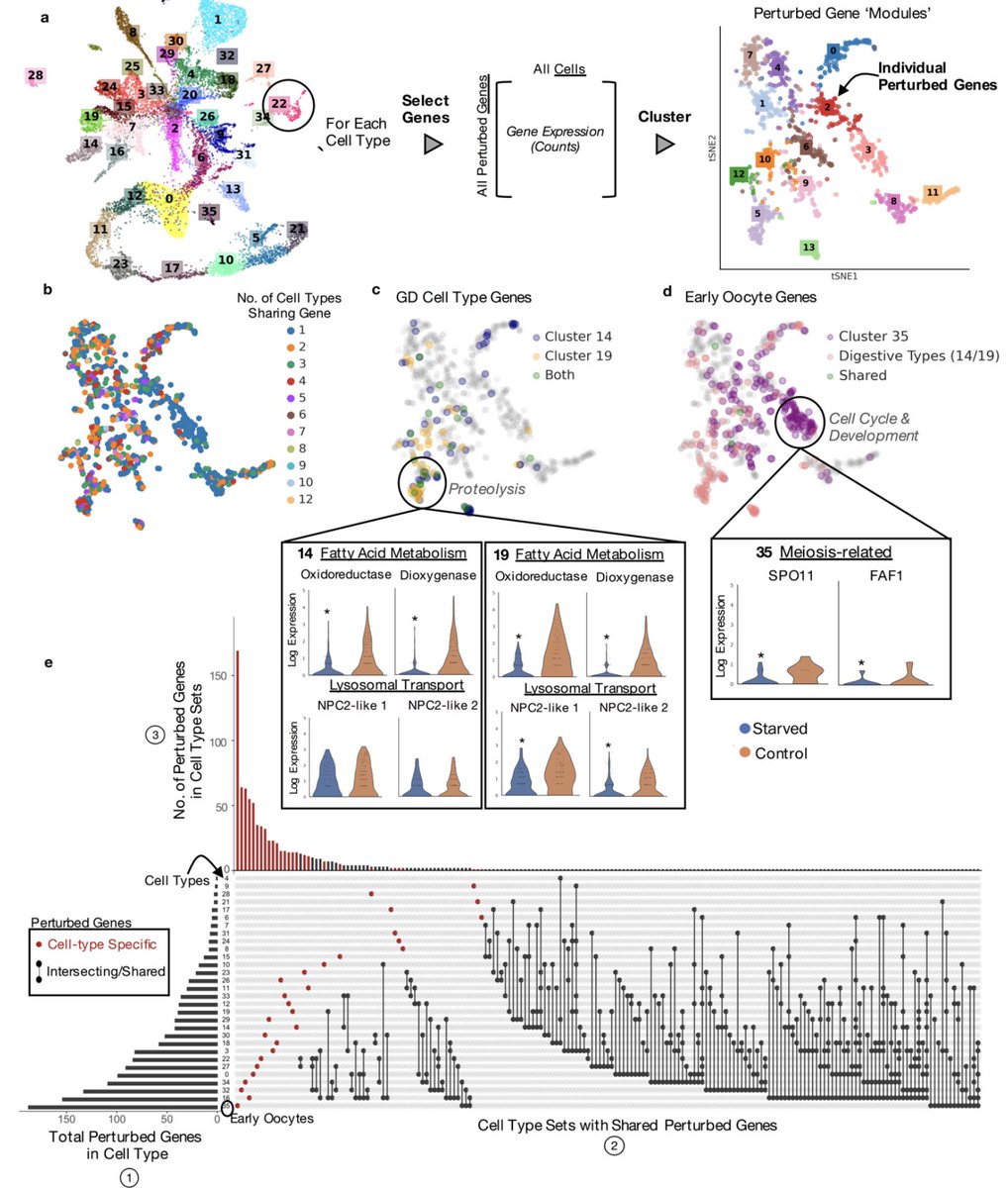
A deep dive into the genes perturbed by starvation was extremely interesting. For example we found one "gene module" of perturbed genes shared across multiple gastro-digestive cell types that was enriched in proteolytic genes. Much more on this in the paper. 9/
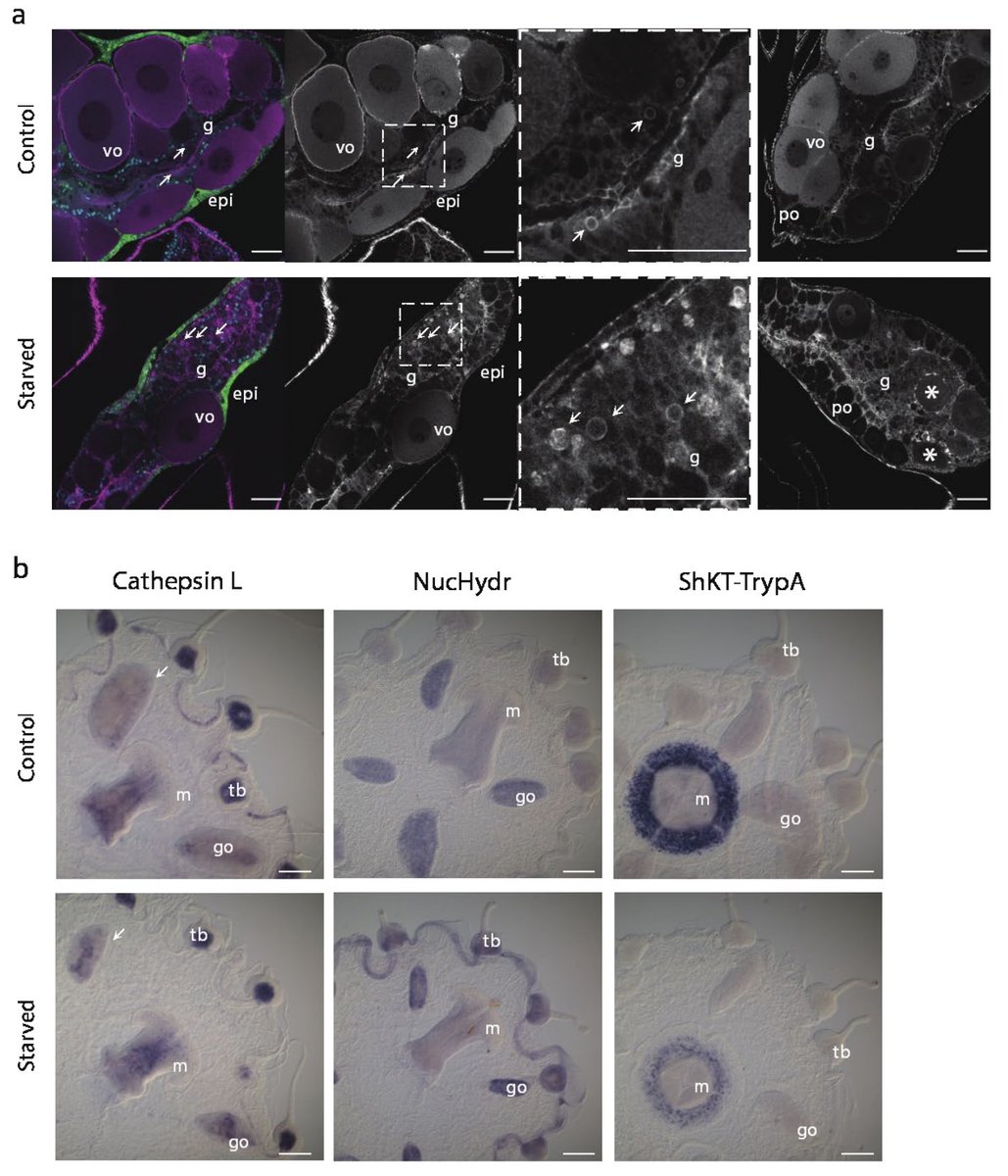
In situs of perturbed genes was illuminating (beautiful work by @AnnaFerraioli_ @Leclere_L @Clytia_Vlfr). E.g., the gastro-digestive cell marker CathepsinL confirms extensive reorganization of the gastroderm in starved animals, especially in the oocyte-depleted gonad (arrows).10/
We have released all the data & code needed to reproduce the paper, including the reads. This reproducibility is not theoretical: every figure caption has a link to a @GoogleColab notebook enabling free reproducibility in the cloud. See e.g. Figure 2: github.com/pachterlab/CWG… 11/
One of our goals was to demonstrate that WHAM-seq can be practiced routinely. The lab cost of our starvation experiment was ~$12,000, which is not a trivial amount but hopefully in the range of most labs. In addition to usable and reproducible code all protocols are shared. 12/
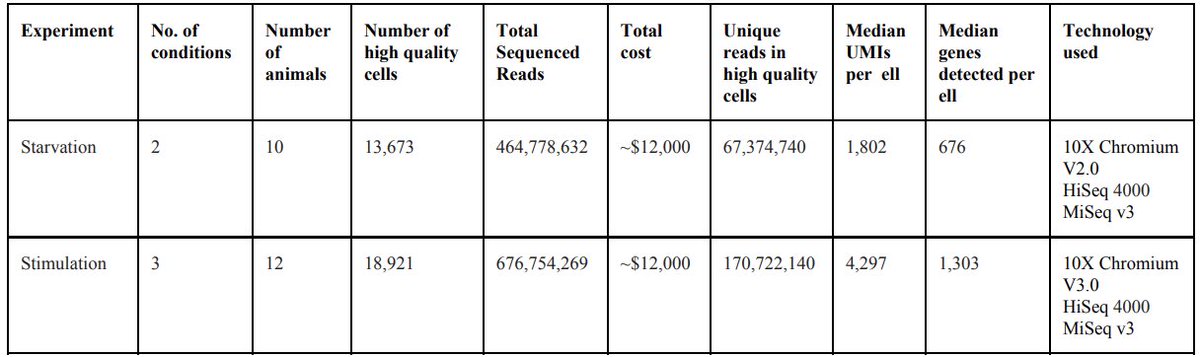
WHAM-seq should be useful for studying developing embryos to organoids to non-models organisms. It is also well suited to large-scale perturbation studies. Clytia hemisphaerica with ~10^5 cells was the perfect scale for now. More will be possible soon. academic.oup.com/database/artic… 13/
Working on this project was a lot of fun. Our author contribution description "T.C., B.W.,J.G., A.F., L.L., R.R.C., E.H., D.J.A, and L.P. contributed to writing and editing the manuscript" reflects to a wonderful, productive collaborative process with tons of discussion. 14/
This was not a project from my lab or the Anderson lab or the Houliston lab. Everyone pitched in key ideas, contributions and experiments for positive epistasis. I've learned a lot about Clytia hemisphaerica from @Clytia_Vlfr & Brady Weissbourd, and I'm now totally hooked... 15/
Special shout out to the leading authors: my former student @JaseGehring did great work w/ the #scRNAseq in our lab (now a postdoc with @JShendure), current student Tara Chari led the analysis, and Brady Weissbourd introduced us to Clytia and guided the neuronal work. 🎐16/16
• • •
Missing some Tweet in this thread? You can try to
force a refresh





