Say you want to study how a protein works... What can you do? A thread on recombinant protein expression and purification (with links to more) 1/
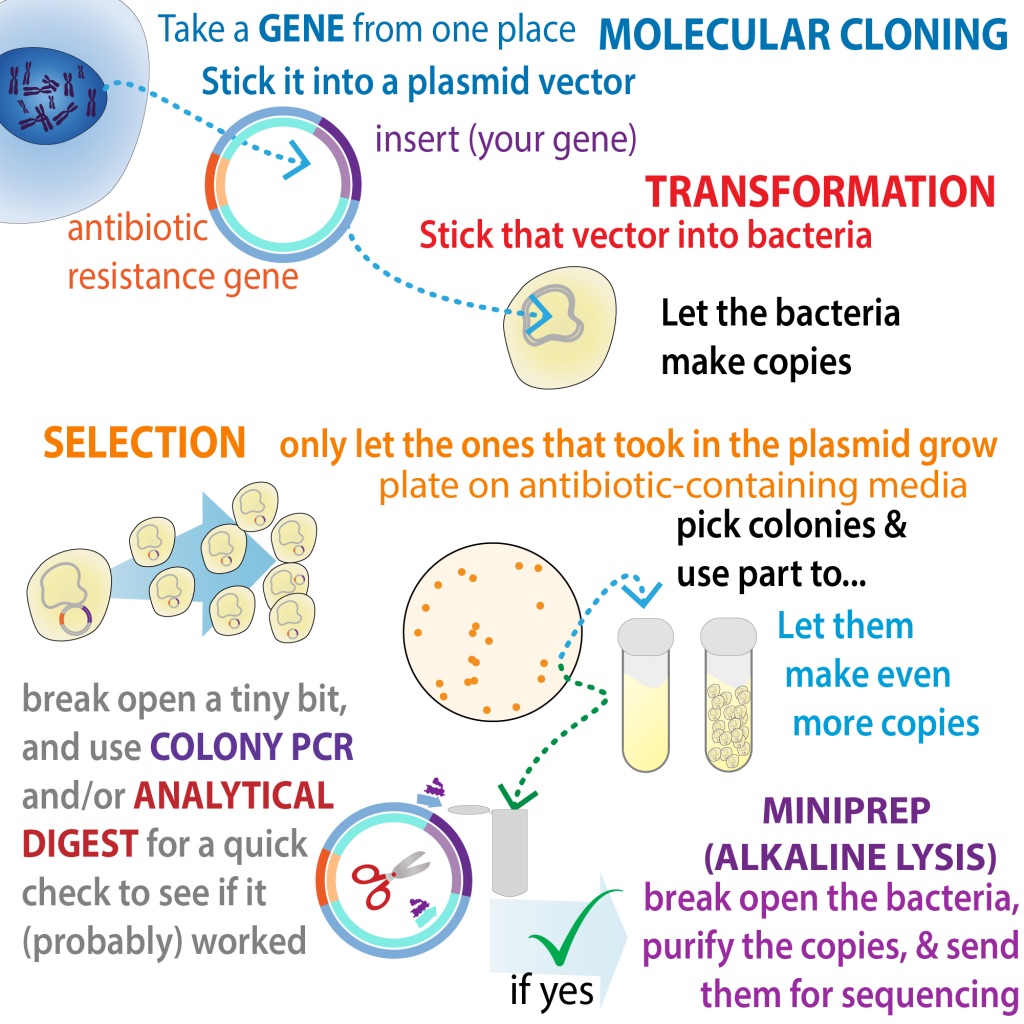
The first step's molecular cloning, where you take the genetic instructions for making that protein (cDNA) & stick them into another piece of DNA (such as a plasmid) that you can use as a "vector" (vehicle) to get the instructions into expression cells bit.ly/molecularcloni… 2/ 

A common form of molecular cloning uses restriction enzymes to "cut and paste" the DNA from one place to another 3/ bit.ly/restrictionenz…

Other methods, such as SLIC, use PCR to make lots of copies of each part you want to join, adding on matching overlapping ends, then mix them together and let bacteria match them together 4/ bit.ly/molecularcloni…

Regardless of how you made your plasmid, you now want to stick it into bacteria cells so they'll make lots of copies of it. We call "sticking genetic info into bacterial cells" transformation & a common way is heat shock with chemically competent cells 5/ bit.ly/transfectionme…

In addition to the insert you put in, the plasmid usually has an antibiotic resistance gene you can use to "select" for bacteria that took in the plasmid 6/ bit.ly/antibioticsele…

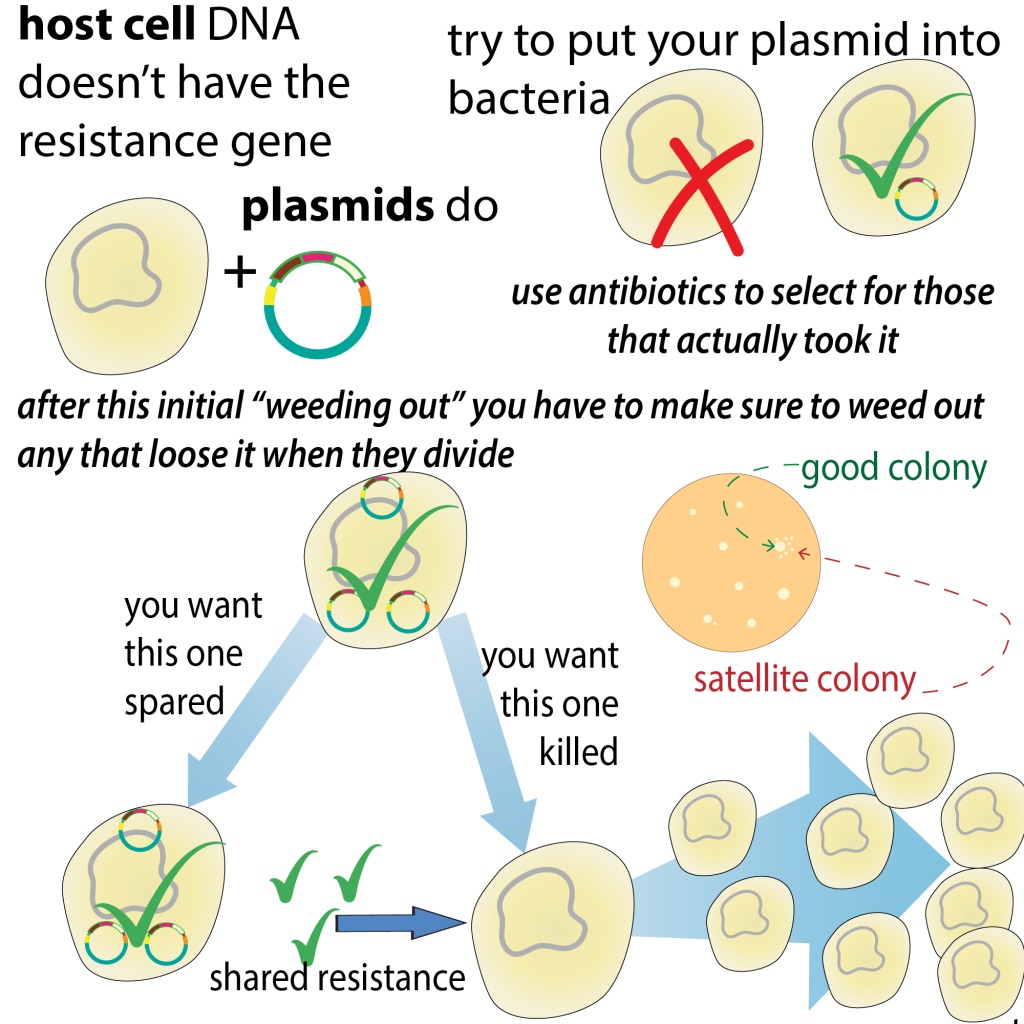
So, the bacteria that survive (and grow as globs on your agar plate called colonies) have the plasmid. But do those plasmids actually have the insert you want? You can check this with an analytical digest or colony PCR 7/ bit.ly/colonychecking


Those can tell you if the insert is there, but is everything spelled right? Were mutations introduced during the cloning process? For that you need to send it for sequencing 8/ bit.ly/colonypcretc

But first you'll need to purify it out of the bacteria which you can do with a MiniPrep kit (or alternative version of alkaline lysis). 9/ bit.ly/minipreps

Once you verify all's okay, you want to stick the plasmid (that has the protein instructions - yay!) into expression cells. The cells you used during the cloning part were great for making lots of plasmid DNA. But now you want cells that are great for making lots of protein 10/
For some proteins, bacteria (a different strain) are good for this (and cheapest). And you can use an inducible T7 promoter to get them to make LOTS of your protein on demand 11/ bit.ly/bacoverexpress…

Other proteins are trickier though (if they require folding help or modifications, etc.) so you might need to try more "complex" cells such as yeast, mammalian, or, what I use, insect cells. 12/ bit.ly/bevsinsect


So, you've gotten some sort of cell to express (make) your protein. Now what? First you need to harvest the cells (separate the cells from all the liquid food (media) they were growing in). You do this by centrifuging them (spinning them down fast) to get the cells to pellet 13/
You can then pour off the liquid part (supernatant), resuspend the pelleted cells in (a smaller volume of) fresh liquid, then freeze them until you're ready to purify them. And then go wash those flasks and bottles... 14/

When you're ready to purify the protein... 1st, you need to break open the cells (lyse them). I use freeze-thaw, high salt, and ultrasonication to help (the sonication also helps shear the DNA (tear it up into pieces) so DNA globs don't get in the way 15/ bit.ly/ultrasonicshea…


Once the cells are broken open, you want to separate the soluble stuff (including your protein unless you're studying a membrane protein or your protein isn't happy) from the insoluble stuff like the membrane bits. Back to the centrifuge, but this time a faster one! 16/

Now we're ready to purify the protein! Here's a roadmap of where we've been and where we're going 17/ bit.ly/proteincleaning

The main way we purify proteins is w/a technique called chromatography. We use columns filled w/little beads (resin) to separate proteins based on how they interact w/beads when you flow the proteins through the column. Different resins separate based on different properties 18/

Sometimes we do this using gravity flow (often in the cold room...) 19/

Other times we use a liquid-handling machine called an FPLC, such as an AKTA to give gravity some help and direct the proteins onto the column at a consistent speed 20/ bit.ly/meettheakta


Often you do a several-step purification, where you use different types of chromatography sequentially, getting your protein purer and purer each time 21/

Often, the first step is a "capture" step where you use affinity chromatography to capture your protein of interest based on something unique about it, such as an affinity tag you added to the end of the protein when you were doing the cloning 22/

A common form of affinity chromatography is a His-Tag and IMAC (Immobilized Metal Affinity Chromatography) such as a Ni-NTA column. 23/ bit.ly/histidineimac

Another affinity chromatography option is a strep tag and streptactin resin 24/ bit.ly/streptag

The affinity step should get rid of most of the other proteins, and now you can cut the tag off if you want - normally you have an endoprotease cleavage site in between the tag and your protein 25/ bit.ly/proteasess

Now your protein is tagless and mostly pure. But you want to get it really pure. Nothing really makes your protein truly unique anymore, so you'll need to separate based on natural differences between proteins... 26/
One such exploitable difference is charge - different proteins have different charges because some amino acids (protein letters) are (sometimes) charged and since different proteins have different spellings, they have different charges 27/

We can take advantage of these charges to separate proteins based on how strongly they stick to a charged resin. We call charged things "ions" so we call this technique ion exchange chromatography 28/ bit.ly/ionexchangechr…


Now your protein is (hopefully) mostly mostly pure, but you want to get it mostly mostly mostly pure so you need to turn to another protein property since at this point, all the proteins in the mix have similar charges.... How about size? 29/
Size exclusion chromatography (SEC), aka gel filtration, separates proteins by their size/shape - the beads have secret tunnels in them only small things can enter. Small proteins thus have a longer path to travel so it takes them longer to get through 30/ bit.ly/sizeexclusionc…


Our protein should now be mostly mostly mostly pure, but how can we know for sure? SDS-PAGE time! This technique separates small amounts of proteins by their length in a thin slab gel and then you can stain the gel and see different proteins as bands 31/ bit.ly/sdspageruler

The purer the protein, the fewer the bands you'll see (although there could be proteins of the same length whose bands overlap...) 32/

You can compare to a ladder of proteins of known size to approximate the size of the proteins in the gel, and see if it matches the protein you were trying to purify. But is it *really* your protein? You can do a western blot to confirm 33/
western blots use antibodies specific to the protein you're looking for as probes to search those protein bands after you transfer them out of the gel and onto a sturdier membrane 34/ bit.ly/westernblotwor…

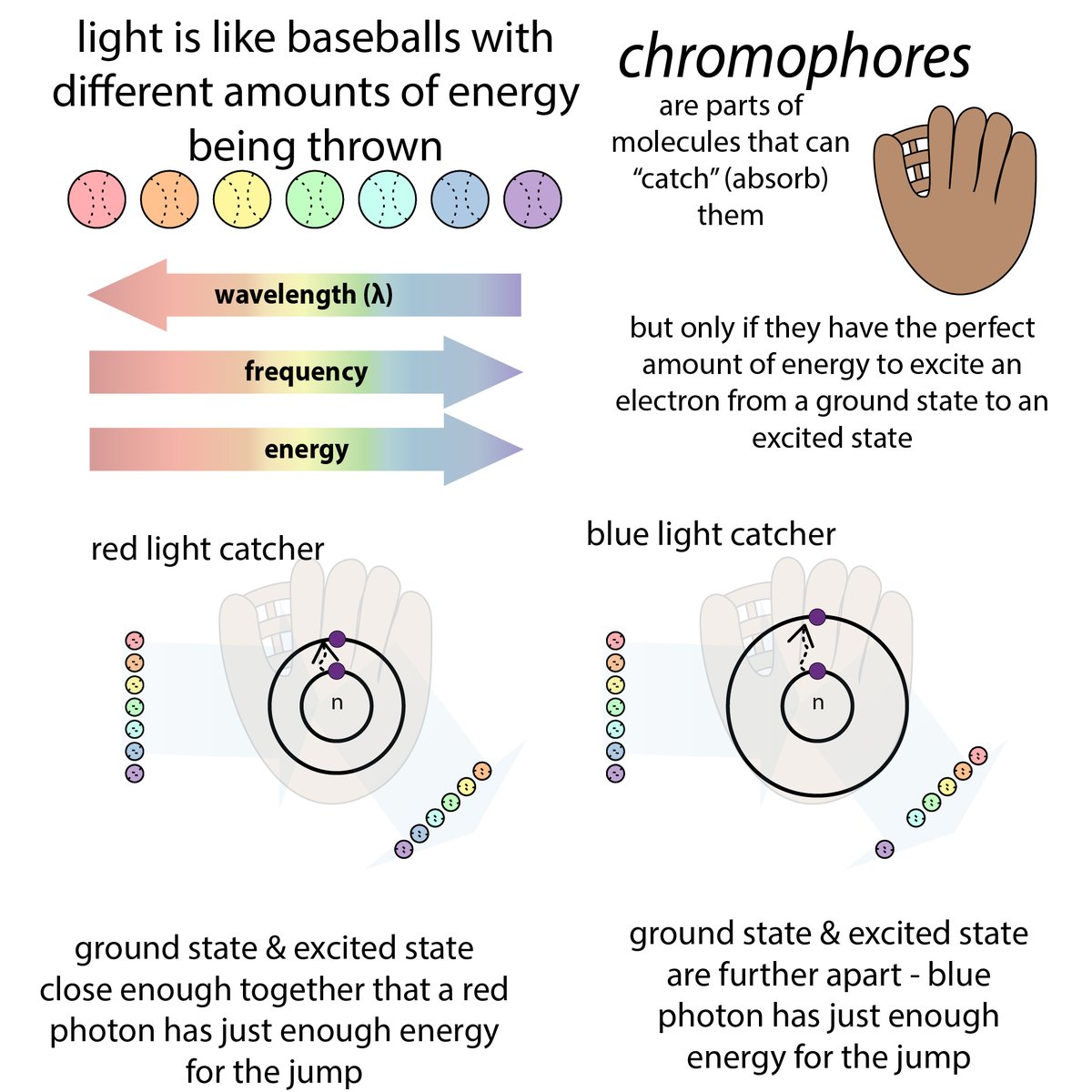
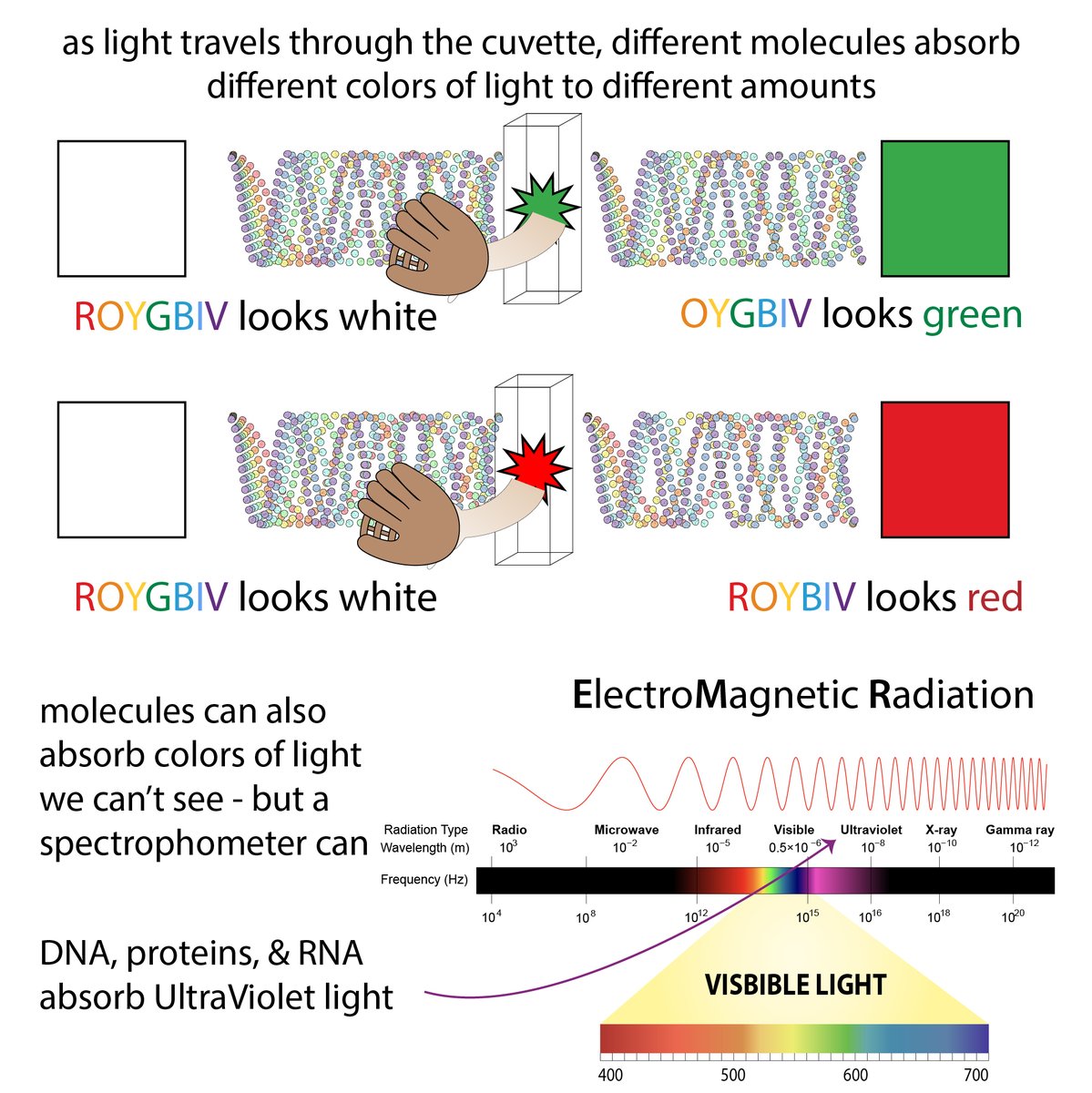
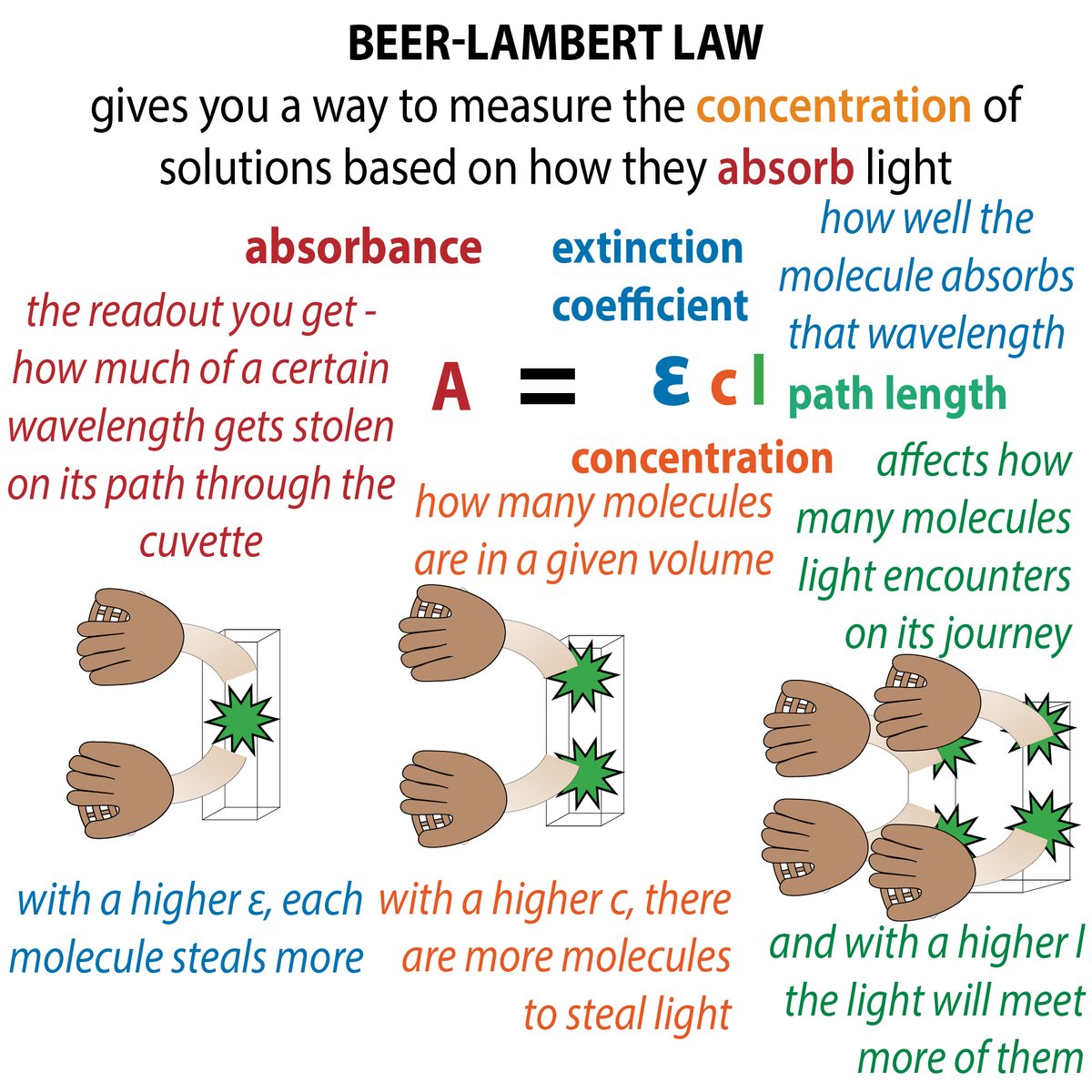
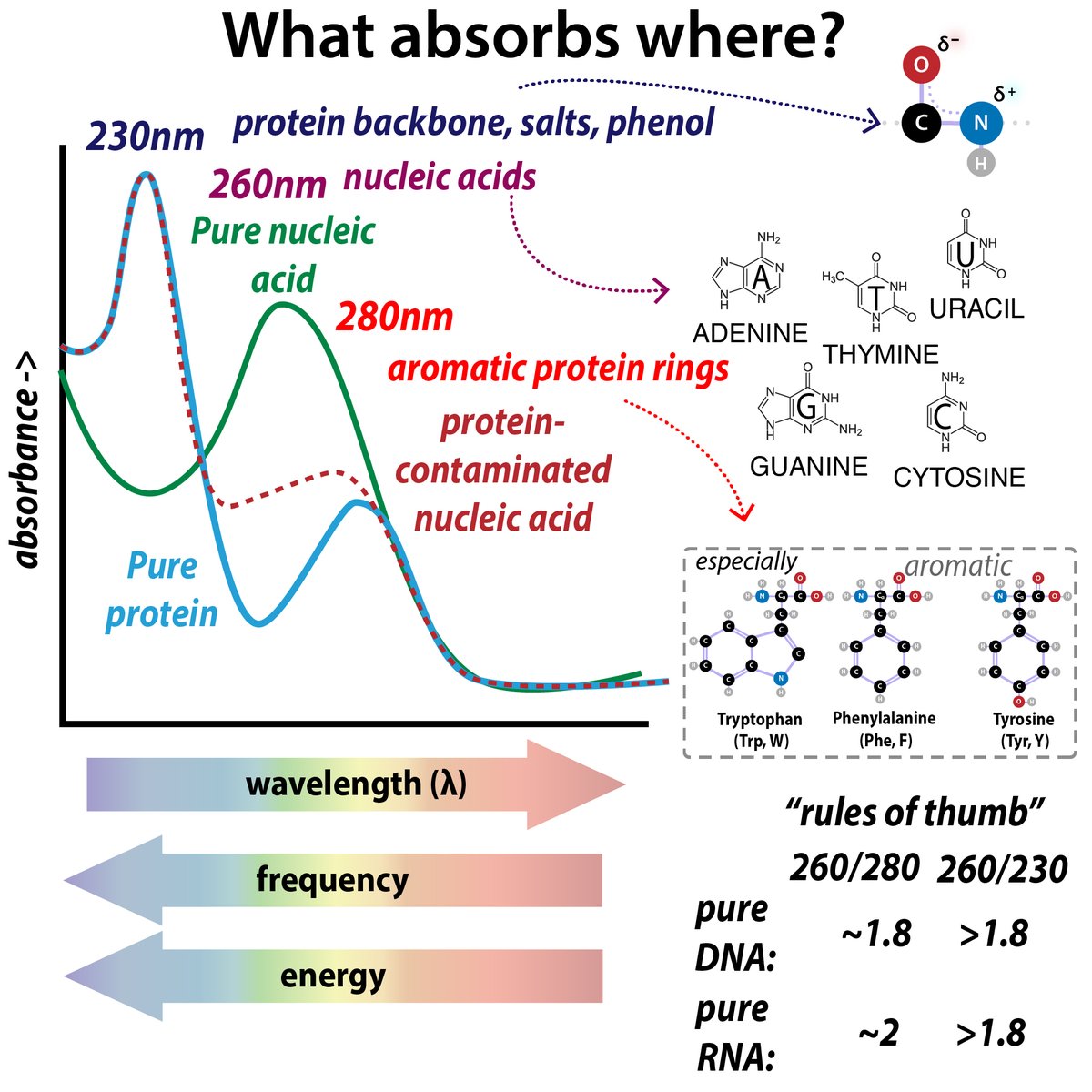
So you've gotten pure protein. Yay! How much? There are a variety of methods for measuring protein concentration, each with pros and cons 35/ bit.ly/proteinmeasuri…

Speaking of concentration, you usually need to concentrate your protein, which you can do w/centrifuge-based concentrators that have membranes w/holes that keep your protein but let excess liquid through (centrifugal ultrafiltration) 36/ bit.ly/proteinconcent…


For long-term storage, you're gonna wanna keep your protein in the super cold (-80°C) freezer - those ones that some vaccines need to be kept in 37/

But you don't want water to freeze in your protein and damage them, so you take a couple steps to prevent ice formation. One is to add a cryoprotectant ("antifreeze") like glycerol that breaks up water networks 38/ bit.ly/cryoprotectant…

Another is to freeze it really really quickly so water doesn't have time to form the intricate network of a crystal lattice. We can do this "flash freezing" using liquid nitrogen. 39/ thebumblingbiochemist.com/365-days-of-sc…


Once your protein's safely stowed - celebrate your success! And get a little rest before the really exciting part - playing with it to figure out how it works! 40/40
@threadreaderapp unroll
• • •
Missing some Tweet in this thread? You can try to
force a refresh