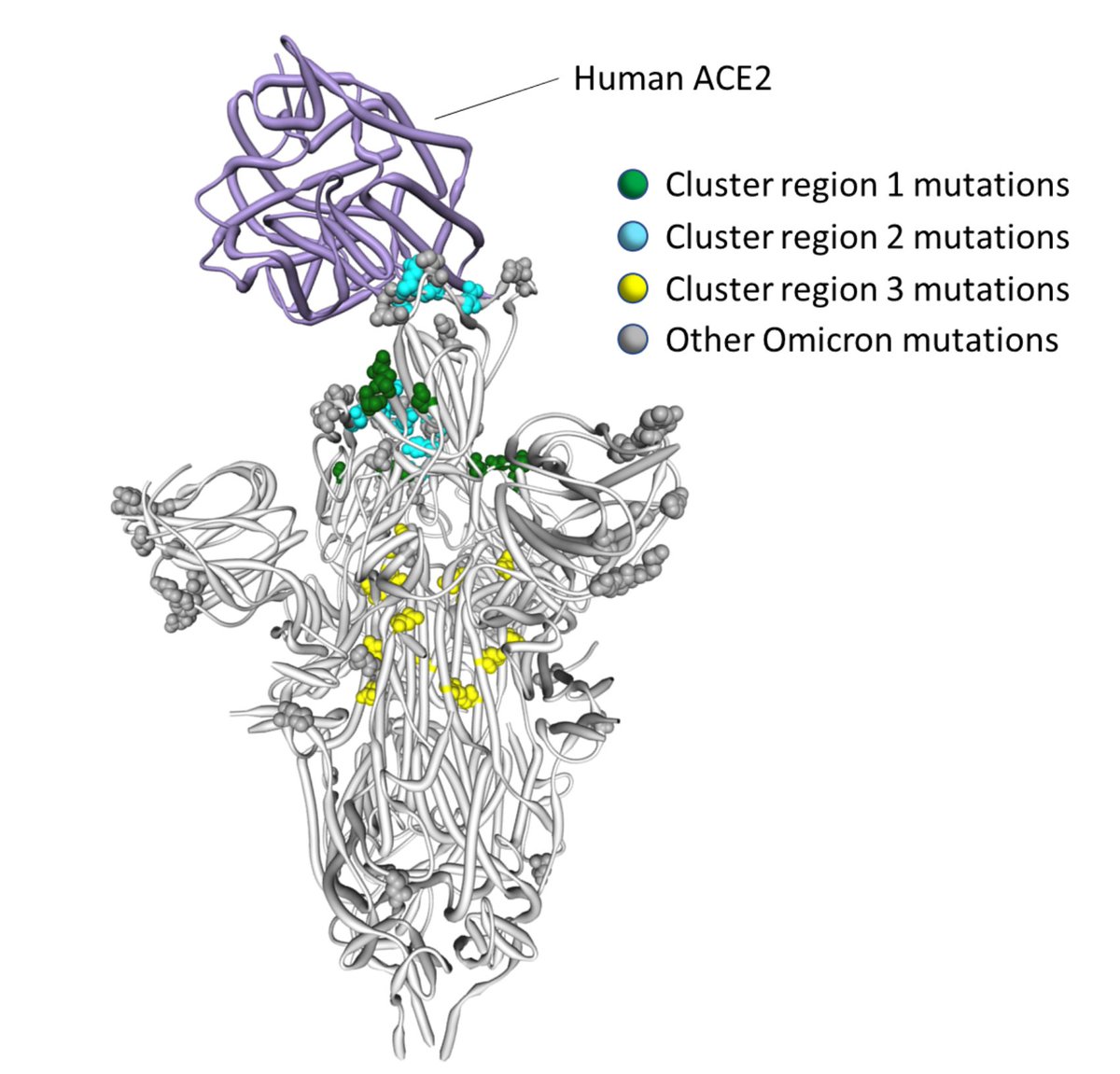
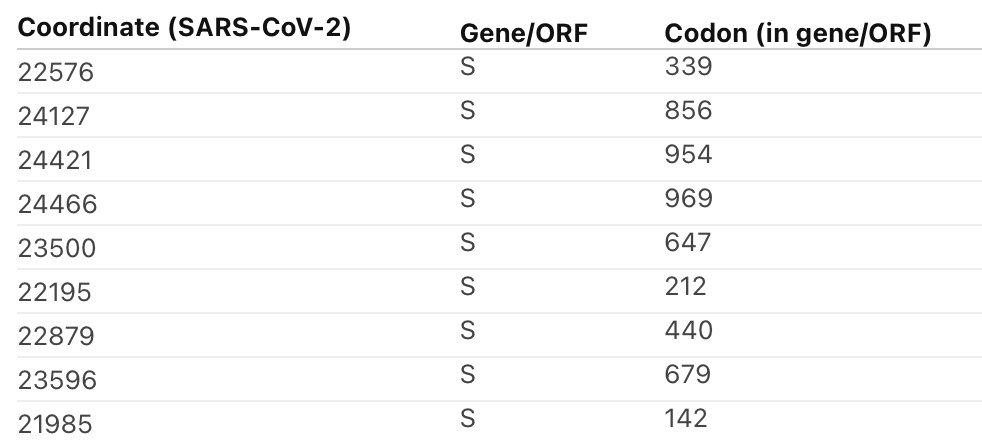
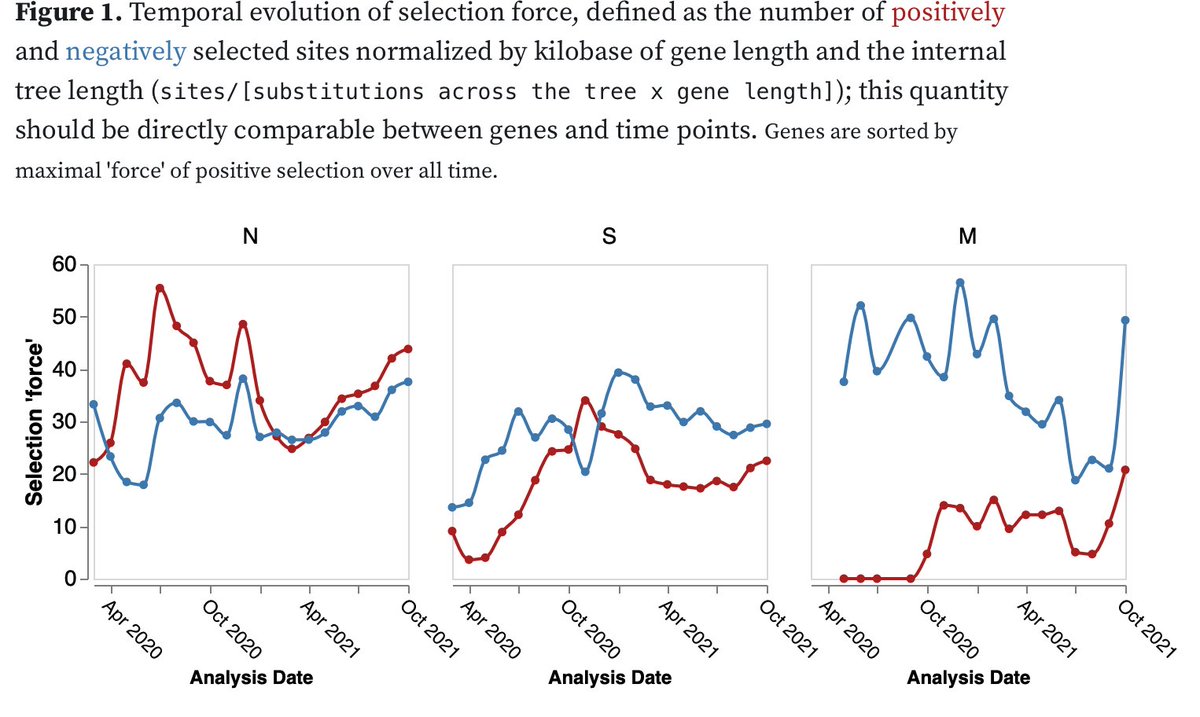
1/4 In a May 2021 preprint on the common evolutionary trajectories of human beta-coronaviruses, we (@EvolveDotZoo, Marina Escalera-Zamudio and others) identified four sites, including S/796 found in #omicron that we hypothesized might be involved in human adaptation 

2/4 In particular, S/796 has experienced what we termed "stepwise evolution" in SARS-CoV-1 and is near the trimerization surface, which undergoes conformational rearrangements during viral fusion

3/4 These types of comparative analyses of beta-coronavirus evolution in humans are effectively studies of evolutionary "replicates" where commonalities commonalities among different viruses can be exploited to develop predictions of what similar viruses may do.
4/4 If we want to learn more about #SARS_CoV_2 (or any other important human pathogen), it always pays to sequence and study similar viruses, even if they are relatively benign (common cold). Comparative evolution is very powerful. Data and viz at observablehq.com/@spond/beta-co…
• • •
Missing some Tweet in this thread? You can try to
force a refresh