1/18 "Selection analysis identifies significant mutational changes in Omicron that are likely to influence both antibody neutralization and Spike function" virological.org/t/selection-an… @DarrenM98230782 @Tuliodna @jbloom_lab @robertson_lab @nekrut @LemeyLab and many others.
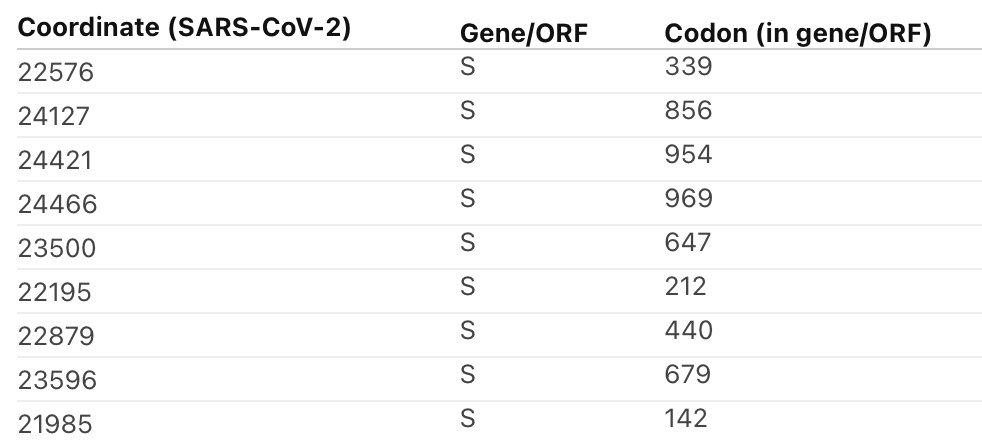
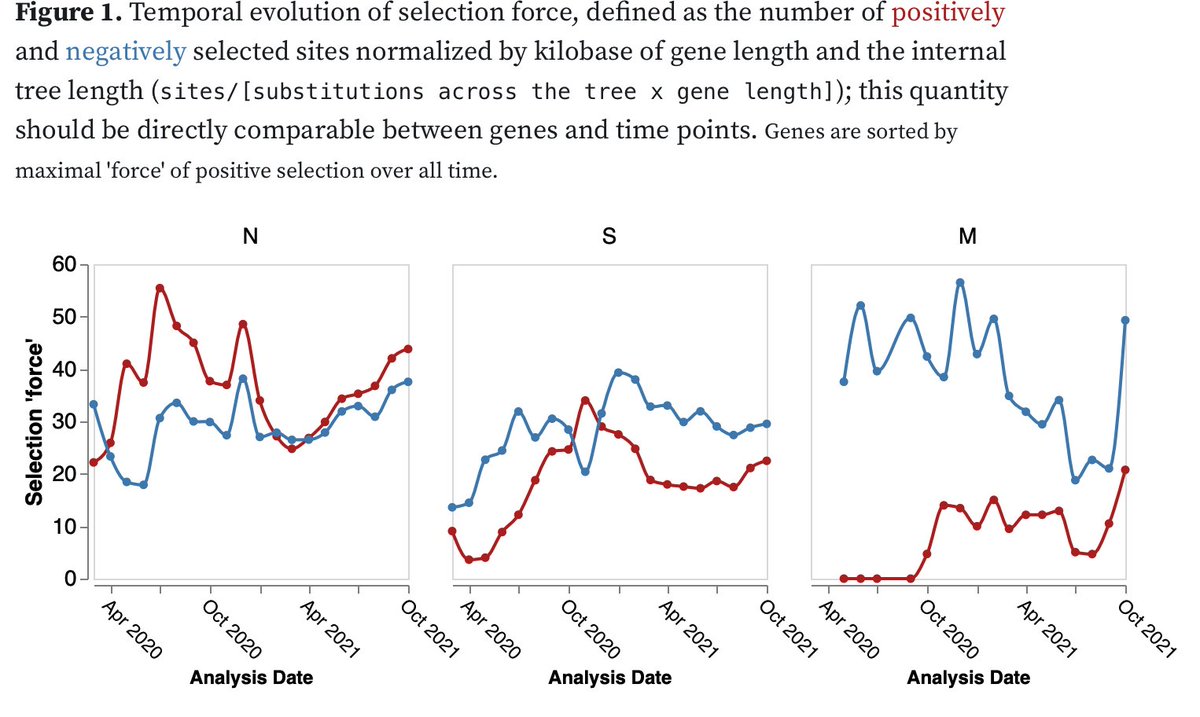
2/18 There are 14 mutation in #omicron S-gene that have been under negative selection (or neutral evolution) prior to Nov 2021. This pattern was NOT seen in previous VOC where many of the signatures sites had been detectably selected prior to emergence cell.com/cell/pdf/S0092…
3/18 The 14 #Omicron mutations fall into 3 clusters in Spike 
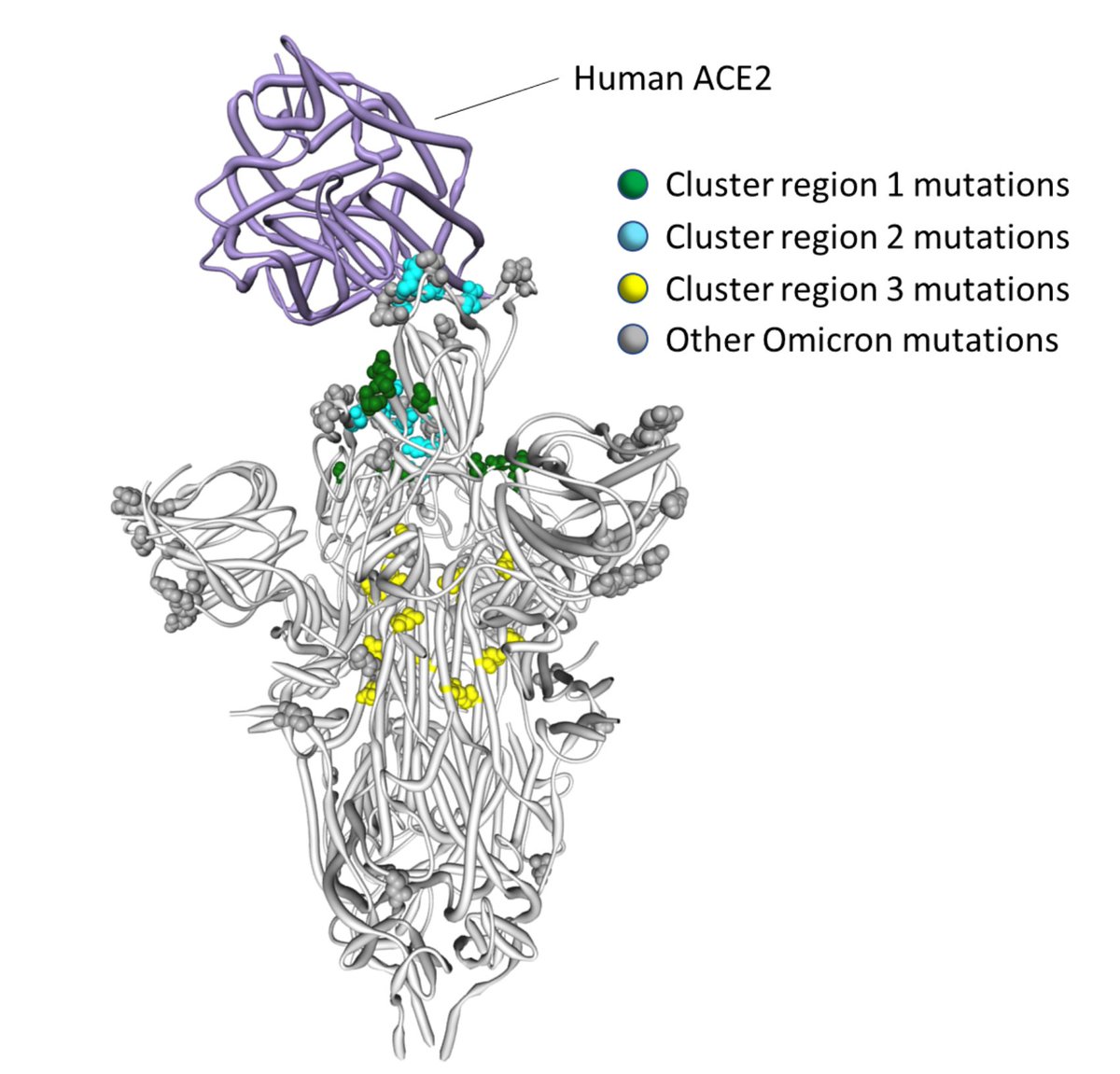
4/18 Cluster region 1 in the RBD: codons/amino acids S/339, S/371, S/373 and S/375; may be targeted by some class 4 neutralizing antibodies
5/18 Cluster region 2 in the RBM including codons/amino acids S/493, S/496, S/498, and S/505. This region is known to be targeted by class 1 and class 2 neutralizing antibodies. S/493 is a known target of such antibodies and has been selected in vitro
6/18 Cluster region 3 in the fusion domain: codons/amino acids S/764, S/856, S/954, S/969, S/981; a region of Spike not known to be currently targeted by neutralizing antibodies
7/18 These mutations are **individually** likely maladaptive: they occur intra-host, but are selected against in population genomes, they occur at cites generally conserved in sarbecoviruses, and they do not co-occur more frequently that expected by chance in other lineages.
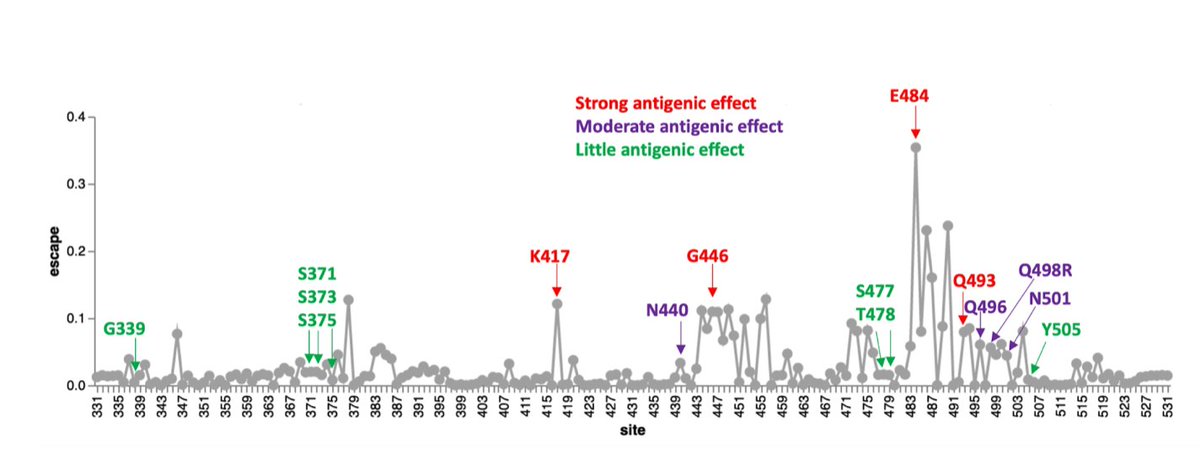
8/18 Yet given the spread and competitive profile of #Omicron, they are **not** maladaptive together, and likely -- adaptive. Notably, only a few of these mutations are expected to have a significant effect on their own antigenically (DMS data from @jbloom_lab)
9/18 So, how and why have so many apparently maladaptive mutations been assembled within Omicron?
Omicron progenitor accumulated its unprecedented number of mutations during an extensive time period of undetected replication.
Omicron progenitor accumulated its unprecedented number of mutations during an extensive time period of undetected replication.
10/18 There are several plausible routes discussed elsewhere: undetected circulation in low surveillance areas, chronic intra-host evolution, reverse zoonosis. More data are needed to resolve which one (could also be a combination of these routes).
11/18 Relative to evolution during normal SARS-CoV-2 person-to-person transmission, evolution within the context of either long-term infections or an alternative animal host could potentially have occurred at an accelerated pace
12/18 In these contexts purifying selection may have been relaxed: enough so for genomes carrying suboptimal combinations of epistatically interacting mutations to remain viable while fitter combinations were discovered via additional mutations and genetic recombination.
13/18 In addition chronic infections are not impacted by the tight transmission bottlenecks that can stochastically purge nascent adaptive mutations during normal transmission.
14/18 Sequential cycles of immune surveillance and viral immune escape within a long-term infection could also potentially explain the mutation clusters without the need to invoke compensatory epistatic interactions between mutations.
15/18 The clustered mutation patterns in the Omicron Spike are reminiscent of those seen in the HIV envelope protein as a consequence of sequentially acquired virus mutations that evade the progressively broadening neutralization potential of a maturing antibody lineage
16/18 As #omicron spreads among human hosts, any deleterious (at the population level) mutations it has retained are expected to revert (possibly slowly, depends on the selection coefficient which could be low).
17/18 OTOH, if the rarely seen mutations in #Omicron show no signs of reverting, rather than supporting one origin hypothesis over another, it would support the hypothesis that these mutations are broadly adaptive when they occur in the combinations found in #Omicron
18/18 Rather than just small tweaks in the antigenicity of Spike, its ACE2 binding properties or its membrane fusion functions, the clustered "rarely seen" mutations in Omicron’s RBD and fusion domain could cause quite big shifts in the way that Spike works
• • •
Missing some Tweet in this thread? You can try to
force a refresh