
*Tamanaviruses* - a primer (medium thread)
(1) Tamana bat virus (TABV) is a highly divergent #flavivirus that was isolated in 1974, from a bat trapped at the exit of Tamana cave, on the North slope of Trinidad's Mount Tamana.
(1) Tamana bat virus (TABV) is a highly divergent #flavivirus that was isolated in 1974, from a bat trapped at the exit of Tamana cave, on the North slope of Trinidad's Mount Tamana.

(2) TABV was identified at the Trinidad Regional #Virus Laboratory (TRVL) as part of a post-war virus discovery effort lead by the Rockefeller Foundation.
The #Tamana cave system comprises several large limestone caverns and is a roost for several species of bat.
The #Tamana cave system comprises several large limestone caverns and is a roost for several species of bat.

(3) Millions of bats fly out of Tamana cave exits to feed at dusk, making for an impressive spectacle.
Researchers at TRVL trapped #bats at cave exits, took samples from these bats and attempted to cultivate viruses.
Researchers at TRVL trapped #bats at cave exits, took samples from these bats and attempted to cultivate viruses.

(4) TABV was isolated from a moustached bat (Pteronotus parnellii).
Serological analyses performed at the time indicated that TABV was related to flaviviruses like yellow fever virus (YFV), but only distantly.
doi.org/10.4269/ajtmh.…
Serological analyses performed at the time indicated that TABV was related to flaviviruses like yellow fever virus (YFV), but only distantly.
doi.org/10.4269/ajtmh.…
(5) TABV remained an enigma for decades.
In 1998 Kuno published electron micrographs of TABV propagated in Vero cells showing virus particles with the typical size and morphology of flaviviruses, but no virus sequences could be amplified via PCR.
doi.org/10.1128/JVI.72…
In 1998 Kuno published electron micrographs of TABV propagated in Vero cells showing virus particles with the typical size and morphology of flaviviruses, but no virus sequences could be amplified via PCR.
doi.org/10.1128/JVI.72…
(6) When the TABV genome sequence was obtained in 2002 it only raised more questions.
Despite being closely related to the genus Flavivirus, and having a generally similar genome organisation, TABV is clearly distinct from other members of the genus.
doi.org/10.1099/0022-1…
Despite being closely related to the genus Flavivirus, and having a generally similar genome organisation, TABV is clearly distinct from other members of the genus.
doi.org/10.1099/0022-1…
(7) TABV is not only distinct in its phylogenetic placement, but also in other signature genomic traits such as nucleotide composition and GC content.
doi.org/10.1128/JVI.00…
doi.org/10.1128/JVI.00…

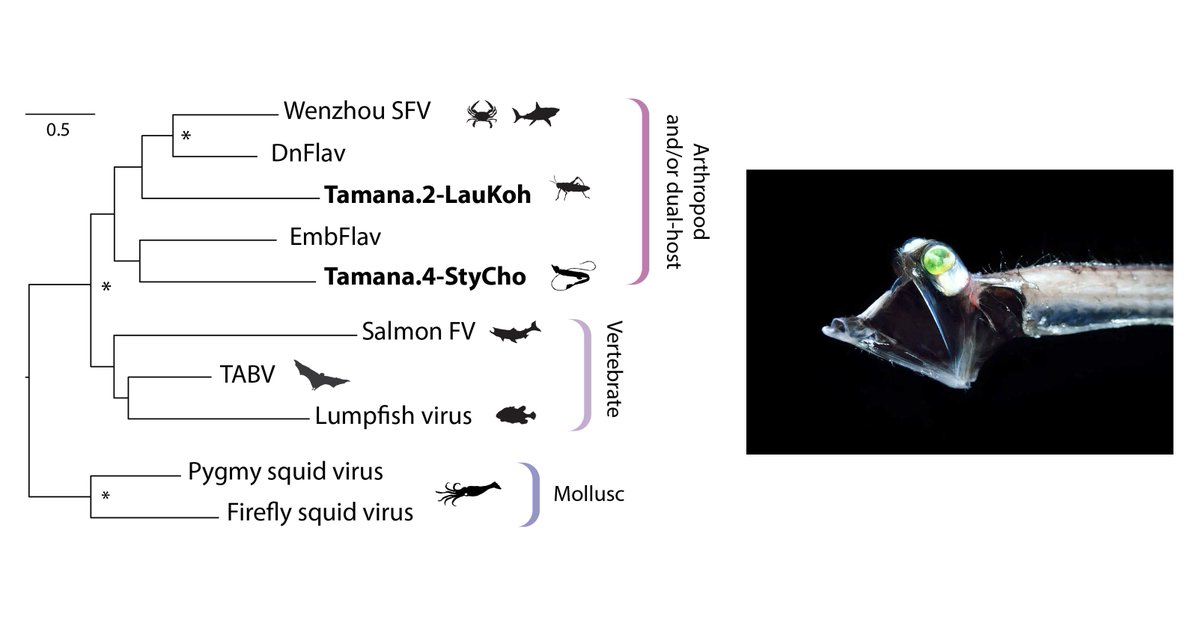
(8) In recent years, however, advances in sequencing technology have lead to the identification of TABV-like viruses in a wide range of animal species, pictured below.
Host species include fish, insects, sharks and squid.
Some of these viruses have been isolated.
Host species include fish, insects, sharks and squid.
Some of these viruses have been isolated.

(9) These findings indicate that TABV is a representative of a novel, distinct group of flaviviruses ('Tamanavirus') that likely infects a broad range of animal species.

(10) In a recent paper, we reported 'genomic fossils' of tamanaviruses in tube-eye fish (Stylephorus), among other species.
This shows that these viruses - like other groups within the Flavivirus family - have ancient origins in the animal kingdom.
doi.org/10.1101/2021.0…
This shows that these viruses - like other groups within the Flavivirus family - have ancient origins in the animal kingdom.
doi.org/10.1101/2021.0…
• • •
Missing some Tweet in this thread? You can try to
force a refresh








