A few folks raised valid geopolitical concerns over this paper firing shots at the Venter Institute/Institut Pasteur SARs-Cov-1 lableak.
This is Beijing claiming one of the premier US genome centers is leaking SARs-CoV-1.
Let's reproduce it.
sciencedirect.com/science/articl…
This is Beijing claiming one of the premier US genome centers is leaking SARs-CoV-1.
Let's reproduce it.
sciencedirect.com/science/articl…
If you want to follow along you will need a Ubuntu instance with a few free software tools installed.
1)Fasterq-dump (Free from NCBI)
2)trim_galore (bioinformatics.babraham.ac.uk/projects/trim_…)
3)samtools (samtools.sourceforge.net)
4)BWA (bio-bwa.sourceforge.net)
5)IGV (Broad Institute)
1)Fasterq-dump (Free from NCBI)
2)trim_galore (bioinformatics.babraham.ac.uk/projects/trim_…)
3)samtools (samtools.sourceforge.net)
4)BWA (bio-bwa.sourceforge.net)
5)IGV (Broad Institute)
First we download the reads they mention in the paper using Fasterq-dump
fasterq-dump ERR1091914
This will download one of the samples in the paper that is listed as being contaminated with SARs-CoV-1.
fasterq-dump ERR1091914
This will download one of the samples in the paper that is listed as being contaminated with SARs-CoV-1.
This sample will be downloaded as a fastq file. This raw sequencing still has illumina primer sequences on them and these need to be trimmed with trim galore. /tools/TrimGalore/trim_galore ERR1091914.fastq
Once you have trimmed reads you can map them to a reference genome or assemble them. Lets stick with read mapping for the moment as assembly requires a bit more software downloads like megahit.
You'll want to download the reference they mention in the paper from NCBI.
You'll want to download the reference they mention in the paper from NCBI.

Click on the genome tab in blue and you will get a .fna file. This will need to indexed with bwa.
bwa mem index SARs-CoV-2_GCF_000864885.1_ViralProj15500_genomic.fna
bwa mem index SARs-CoV-2_GCF_000864885.1_ViralProj15500_genomic.fna
Next step is to map these reads with bwa mem on 4 threads and to pipe this into samtools so you get a .bam file out of the mapping.
bwa mem -t 4 SARs-CoV-2_GCF_000864885.1_ViralProj15500_genomic.fna ERR1091914_trimmed.fq | samtools view -Sb - > ERR1091914.bam
bwa mem -t 4 SARs-CoV-2_GCF_000864885.1_ViralProj15500_genomic.fna ERR1091914_trimmed.fq | samtools view -Sb - > ERR1091914.bam
This bam file just needs to be sorted and indexed and you are ready to load the BAM file into IGV for viewing.
samtools sort ERR1091914.bam -o ERR1091914_sort.bam
samtools index ERR1091914_sort.bam
Copy both of these files to a local drive that has IGV installed.
samtools sort ERR1091914.bam -o ERR1091914_sort.bam
samtools index ERR1091914_sort.bam
Copy both of these files to a local drive that has IGV installed.
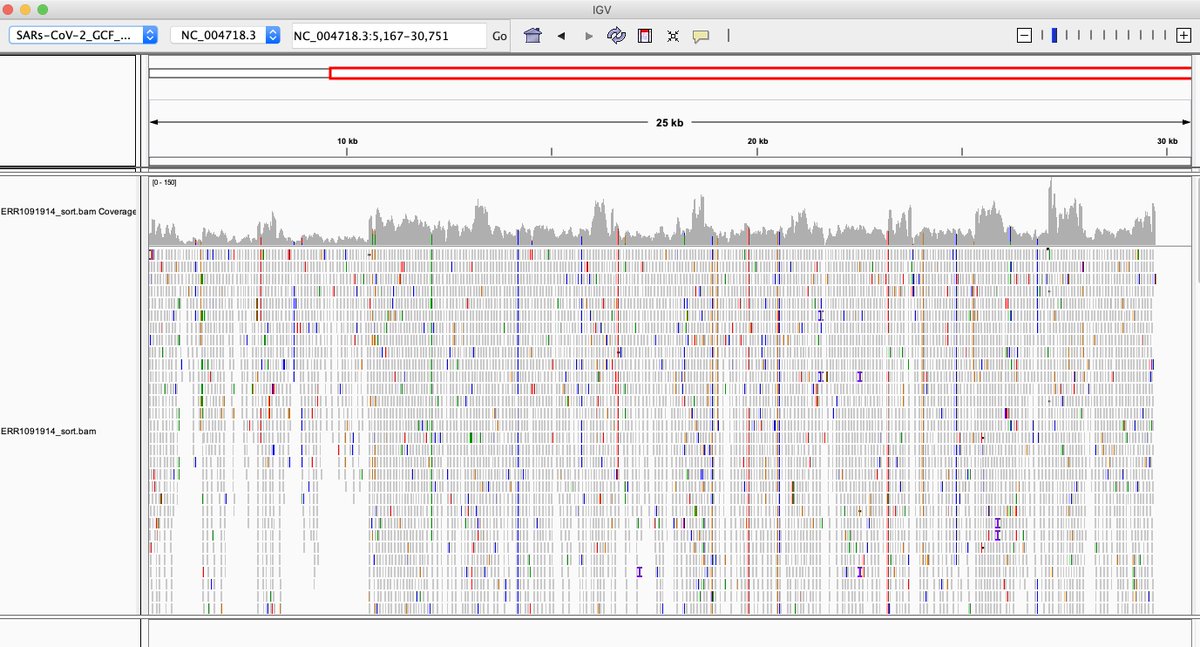
Bingo... There are indeed SARs-CoV-1 sequences in the listed samples in the paper. While it may be from heavily censored China, it is reproducible. ncbi.nlm.nih.gov/sra/?term=ERR1…
• • •
Missing some Tweet in this thread? You can try to
force a refresh