
1/
Should we use #remdesivir (RDV) to treat #COVID19 #SARSCoV2? Let’s take a look at the data.
Here’s: #HowIReadThisPaper on the @NEJM case series of remdesivir for COVID19:
Grein et al: nejm.org/doi/full/10.10…
(Thread)
Should we use #remdesivir (RDV) to treat #COVID19 #SARSCoV2? Let’s take a look at the data.
Here’s: #HowIReadThisPaper on the @NEJM case series of remdesivir for COVID19:
Grein et al: nejm.org/doi/full/10.10…
(Thread)

2/
Background: RDV is an ATP analogue that inhibits many viral RNA polymerases in vitro, including MERS-CoV and SARS-CoV1+2. However, in an RCT of 4 drugs for ebola, RDV was ineffective:
nejm.org/doi/full/10.10…
It does not currently have an FDA-approved indication for use.
Background: RDV is an ATP analogue that inhibits many viral RNA polymerases in vitro, including MERS-CoV and SARS-CoV1+2. However, in an RCT of 4 drugs for ebola, RDV was ineffective:
nejm.org/doi/full/10.10…
It does not currently have an FDA-approved indication for use.
3/
Question: What were the outcomes among patients hospitalized with #COVID19 who received RDV on a compassionate use basis?
Date Published: 10 April 2020
Funding: Gilead Sciences
Question: What were the outcomes among patients hospitalized with #COVID19 who received RDV on a compassionate use basis?
Date Published: 10 April 2020
Funding: Gilead Sciences
4/
Study design: Case series
Population: 61 patients hospitalized in several countries with severe #COVID19 for whom compassionate use of RDV was approved by Gilead
(more on compassionate use, or expanded access: fda.gov/news-events/pu…)
Study period: 1/25/2020 - 3/30/2020
Study design: Case series
Population: 61 patients hospitalized in several countries with severe #COVID19 for whom compassionate use of RDV was approved by Gilead
(more on compassionate use, or expanded access: fda.gov/news-events/pu…)
Study period: 1/25/2020 - 3/30/2020
5/
Intervention: RDV 200 mg IV on day 1 + 100 mg IV daily x9 days (10 days total) in addition to usual care, which varied at each hospital, but at a minimum included non-invasive or invasive oxygen support
Control: none
Intervention: RDV 200 mg IV on day 1 + 100 mg IV daily x9 days (10 days total) in addition to usual care, which varied at each hospital, but at a minimum included non-invasive or invasive oxygen support
Control: none
6/
Study procedures: Treating clinicians submitted an application for RDV to Gilead. Approved patients received a 10-day course, plus usual care as determined by treating clinicians. Clinical and laboratory data were reported daily through day 10, and sporadically thereafter.
Study procedures: Treating clinicians submitted an application for RDV to Gilead. Approved patients received a 10-day course, plus usual care as determined by treating clinicians. Clinical and laboratory data were reported daily through day 10, and sporadically thereafter.
7/
Inclusion criteria: Hospitalized patients with RT-PCR confirmed #SARSCoV2 + SpO2 =< 94% on room air or need for supplemental O2
Exclusion criteria: Renal impairment (CrCl <30 mL/min), active hepatitis (AST or AST >5x ULN), use of any other investigational drug for COVID19
Inclusion criteria: Hospitalized patients with RT-PCR confirmed #SARSCoV2 + SpO2 =< 94% on room air or need for supplemental O2
Exclusion criteria: Renal impairment (CrCl <30 mL/min), active hepatitis (AST or AST >5x ULN), use of any other investigational drug for COVID19
8/
Protocol available? Yes (nejm.org/doi/suppl/10.1…)
Primary outcome: None specified
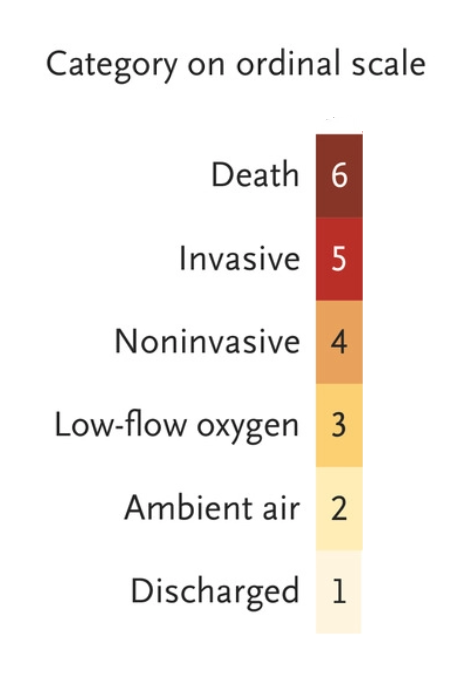
Other outcomes reported: Cumulative incidence of clinical improvement (↓ =>2 levels on scale below or live discharge); change in oxygen support, adverse events, mortality, and lab data
Protocol available? Yes (nejm.org/doi/suppl/10.1…)
Primary outcome: None specified
Other outcomes reported: Cumulative incidence of clinical improvement (↓ =>2 levels on scale below or live discharge); change in oxygen support, adverse events, mortality, and lab data
9/
Primary analysis: Descriptive only
Sample size justification: Not provided
Duration of follow-up: 28 days
Loss to follow-up: 8/61 patients (7 patients contributed no post-treatment data; 1 patient received an incorrect dose of RDV).
Primary analysis: Descriptive only
Sample size justification: Not provided
Duration of follow-up: 28 days
Loss to follow-up: 8/61 patients (7 patients contributed no post-treatment data; 1 patient received an incorrect dose of RDV).
10/
Results:
Table 1: Median age = 64, 75% male
40% of patients were from the USA, 40% from Europe or Canada, 20% from Japan
Median Sx duration before receipt of RDV: 12 days
Oxygen support at baseline:
ECMO: 8%
Intubated: 57%
NIPPV: 4%
HFNC: 9%
O2 via NC: 19%
Room air: 4%
Results:
Table 1: Median age = 64, 75% male
40% of patients were from the USA, 40% from Europe or Canada, 20% from Japan
Median Sx duration before receipt of RDV: 12 days
Oxygen support at baseline:
ECMO: 8%
Intubated: 57%
NIPPV: 4%
HFNC: 9%
O2 via NC: 19%
Room air: 4%

11/
Figure 1: At a median of 18 days [IQR 13-23] after starting RDV, 36 of 53 (68%) patients improved; 8 of 52 (15%) worsened.
All patients who were on room air or low-flow O2 at baseline improved; all who worsened were receiving HFNC, NIPPV, or were intubated at baseline.
Figure 1: At a median of 18 days [IQR 13-23] after starting RDV, 36 of 53 (68%) patients improved; 8 of 52 (15%) worsened.
All patients who were on room air or low-flow O2 at baseline improved; all who worsened were receiving HFNC, NIPPV, or were intubated at baseline.

12/
Figure 2: By day 28:
25 of 53 (47%) patients were discharged
12 of 53 (23%) had improvement in O2 support required
8 of 53 (15%) were unchanged
1 of 53 (2%) worsened
7 of 53 (13%) died
Figure 2: By day 28:
25 of 53 (47%) patients were discharged
12 of 53 (23%) had improvement in O2 support required
8 of 53 (15%) were unchanged
1 of 53 (2%) worsened
7 of 53 (13%) died

13/
Figure 3: The cumulative incidence of clinical improvement at day 28 was 84% [95% CI 70-99%]. Older patients and patients requiring invasive ventilation at baseline improved less often than younger patients and patients not requiring invasive ventilation at baseline.
Figure 3: The cumulative incidence of clinical improvement at day 28 was 84% [95% CI 70-99%]. Older patients and patients requiring invasive ventilation at baseline improved less often than younger patients and patients not requiring invasive ventilation at baseline.

14/
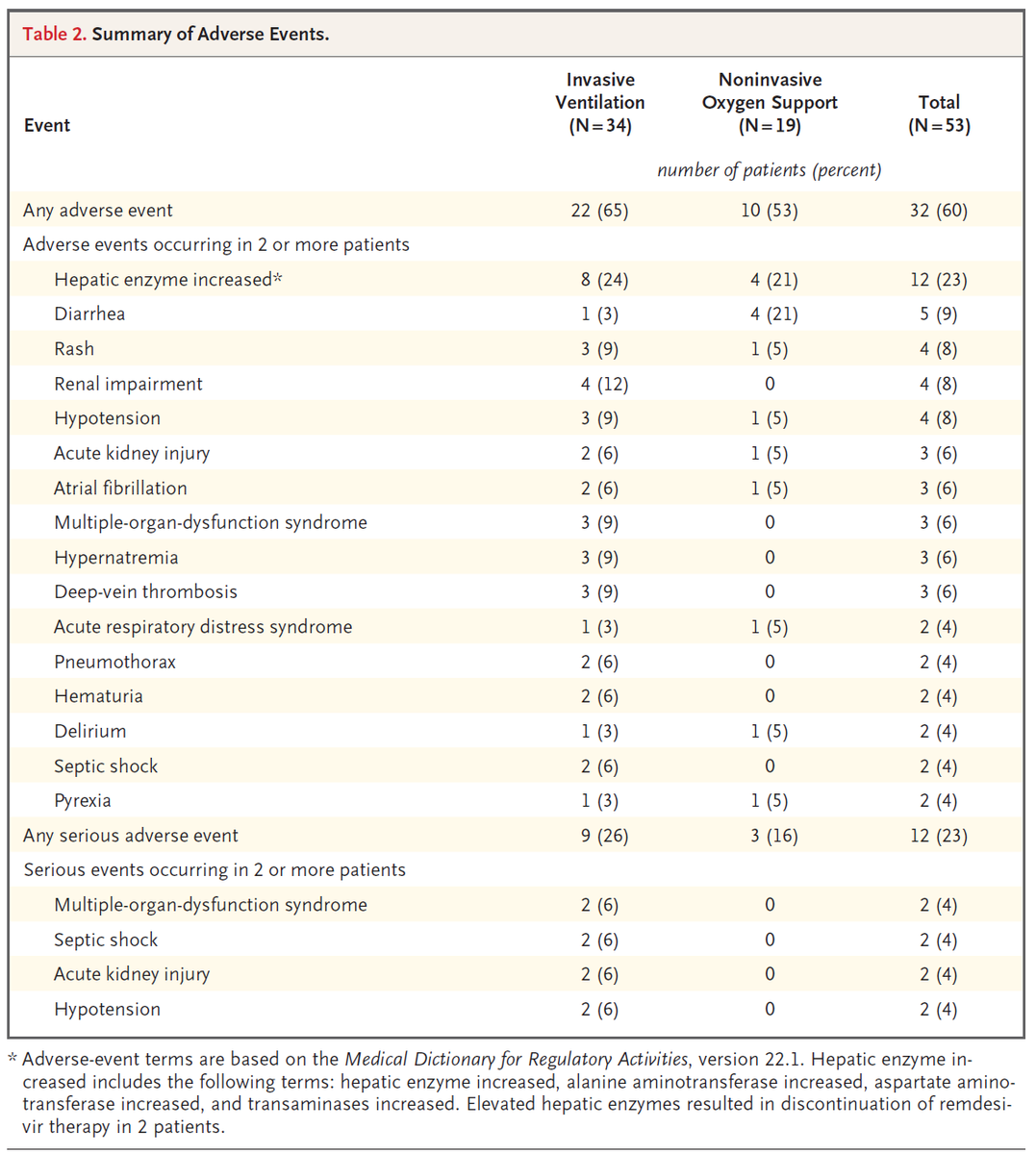
Table 2: 32 of 53 (60%) patients experienced adverse events; 12 of 53 (23%) were serious.
The most common were transaminitis, diarrhea, rash, and AKI.
4 of 53 (8%) of patients stopped RDV early.
Table 2: 32 of 53 (60%) patients experienced adverse events; 12 of 53 (23%) were serious.
The most common were transaminitis, diarrhea, rash, and AKI.
4 of 53 (8%) of patients stopped RDV early.
15/
Author’s conclusions: Despite the lack of a control group, comparing the observed mortality rate of 13% in this severely ill patient group with that seen in other published reports suggests RDV may have benefits in patients with severe COVID19.
Author’s conclusions: Despite the lack of a control group, comparing the observed mortality rate of 13% in this severely ill patient group with that seen in other published reports suggests RDV may have benefits in patients with severe COVID19.
16/
My appraisal: The authors note small size, short follow-up, missing data, and lack of a control group as the main limitations of this study. Accepting those problems, are there other sources of bias to threaten the suggestion that RDV is promising?
Yes. Let’s take a look.
My appraisal: The authors note small size, short follow-up, missing data, and lack of a control group as the main limitations of this study. Accepting those problems, are there other sources of bias to threaten the suggestion that RDV is promising?
Yes. Let’s take a look.
17/
It is tempting to think that descriptive studies are less vulnerable to bias than analytical studies, but let’s see why this is not so.
Two important questions to ask here are:
1. How did patients get into the study?
2. How did outcomes come out of the study?
It is tempting to think that descriptive studies are less vulnerable to bias than analytical studies, but let’s see why this is not so.
Two important questions to ask here are:
1. How did patients get into the study?
2. How did outcomes come out of the study?
18/
The first question is all about looking for selection bias. For a patient to be in this study:
Clinicians had to apply for RDV (n =?)
Gilead had to approve RDV (n =61)
Patients had to complete the RDV protocol (n =53)
Each step introduces the potential for selection bias.
The first question is all about looking for selection bias. For a patient to be in this study:
Clinicians had to apply for RDV (n =?)
Gilead had to approve RDV (n =61)
Patients had to complete the RDV protocol (n =53)
Each step introduces the potential for selection bias.
19/
If the total number of applications for RDV Gilead received was very large, this raises the risk of selection bias, and these results could be more reflective of who received RDV than RDV itself.
To gauge this, we need the denominator, but this was not reported.
If the total number of applications for RDV Gilead received was very large, this raises the risk of selection bias, and these results could be more reflective of who received RDV than RDV itself.
To gauge this, we need the denominator, but this was not reported.


20/
In addition, patients who receive an expanded access medication are likely to differ from those who do not, and clinicians who submit an application are likely to differ from those who do not.
This makes it difficult to know who to generalize the results of this study to.
In addition, patients who receive an expanded access medication are likely to differ from those who do not, and clinicians who submit an application are likely to differ from those who do not.
This makes it difficult to know who to generalize the results of this study to.
21/
Further, the median time from Sx onset to receipt of RDV was 12 days. This means patients had to survive the first 12 days of COVID19 to be eligible for this study.
An early series from Wuhan reported a median time from Sx onset to ARDS of 8 days:
jamanetwork.com/journals/jama/…
Further, the median time from Sx onset to receipt of RDV was 12 days. This means patients had to survive the first 12 days of COVID19 to be eligible for this study.
An early series from Wuhan reported a median time from Sx onset to ARDS of 8 days:
jamanetwork.com/journals/jama/…
22/
Once patients made it into this study, the lack of a standardized definition of “usual care” is another potential source of bias.
Could interventions other than RDV explain the 13% mortality rate, apparently lower than in other series? We don’t know.
Once patients made it into this study, the lack of a standardized definition of “usual care” is another potential source of bias.
Could interventions other than RDV explain the 13% mortality rate, apparently lower than in other series? We don’t know.
23/
For comparison, the authors cite a mortality rate of 66% among intubated patients in another series:
jamanetwork.com/journals/jamai…
In a subsequent larger series (n=1,591) mortality among ICU patients was 26% (of whom 88% were intubated):
jamanetwork.com/journals/jama/…
For comparison, the authors cite a mortality rate of 66% among intubated patients in another series:
jamanetwork.com/journals/jamai…
In a subsequent larger series (n=1,591) mortality among ICU patients was 26% (of whom 88% were intubated):
jamanetwork.com/journals/jama/…
24/
The 2nd Q in tweet 18 is all about ascertainment bias.
Fig 2 raises concern that patients who were sicker at baseline were less likely to be followed until clinical improvement, discharge, or death.
Of the 8 patients with none of those outcomes reported, all were intubated.
The 2nd Q in tweet 18 is all about ascertainment bias.
Fig 2 raises concern that patients who were sicker at baseline were less likely to be followed until clinical improvement, discharge, or death.
Of the 8 patients with none of those outcomes reported, all were intubated.
25/
The most likely situation is that they were still hospitalized at the end of the study period, but because they did not contribute complete person-time data, we don’t know what happened to them. Because they were sicker, this creates ascertainment bias.
The most likely situation is that they were still hospitalized at the end of the study period, but because they did not contribute complete person-time data, we don’t know what happened to them. Because they were sicker, this creates ascertainment bias.
26/
Regarding adverse events, it is difficult to know whether these are due to COVID19 or RDV. The most informative adverse events are those that prompted discontinuation of RDV, which occurred in 8% of patients.
Regarding adverse events, it is difficult to know whether these are due to COVID19 or RDV. The most informative adverse events are those that prompted discontinuation of RDV, which occurred in 8% of patients.
27/
Bottom line: Without a control group, it is impossible to tell whether the observed outcomes in this selected population are due to RDV. Selection bias limits both comparison to other cohorts and the generalizability of these results. Well-designed RCTs are needed.
(End)
Bottom line: Without a control group, it is impossible to tell whether the observed outcomes in this selected population are due to RDV. Selection bias limits both comparison to other cohorts and the generalizability of these results. Well-designed RCTs are needed.
(End)




