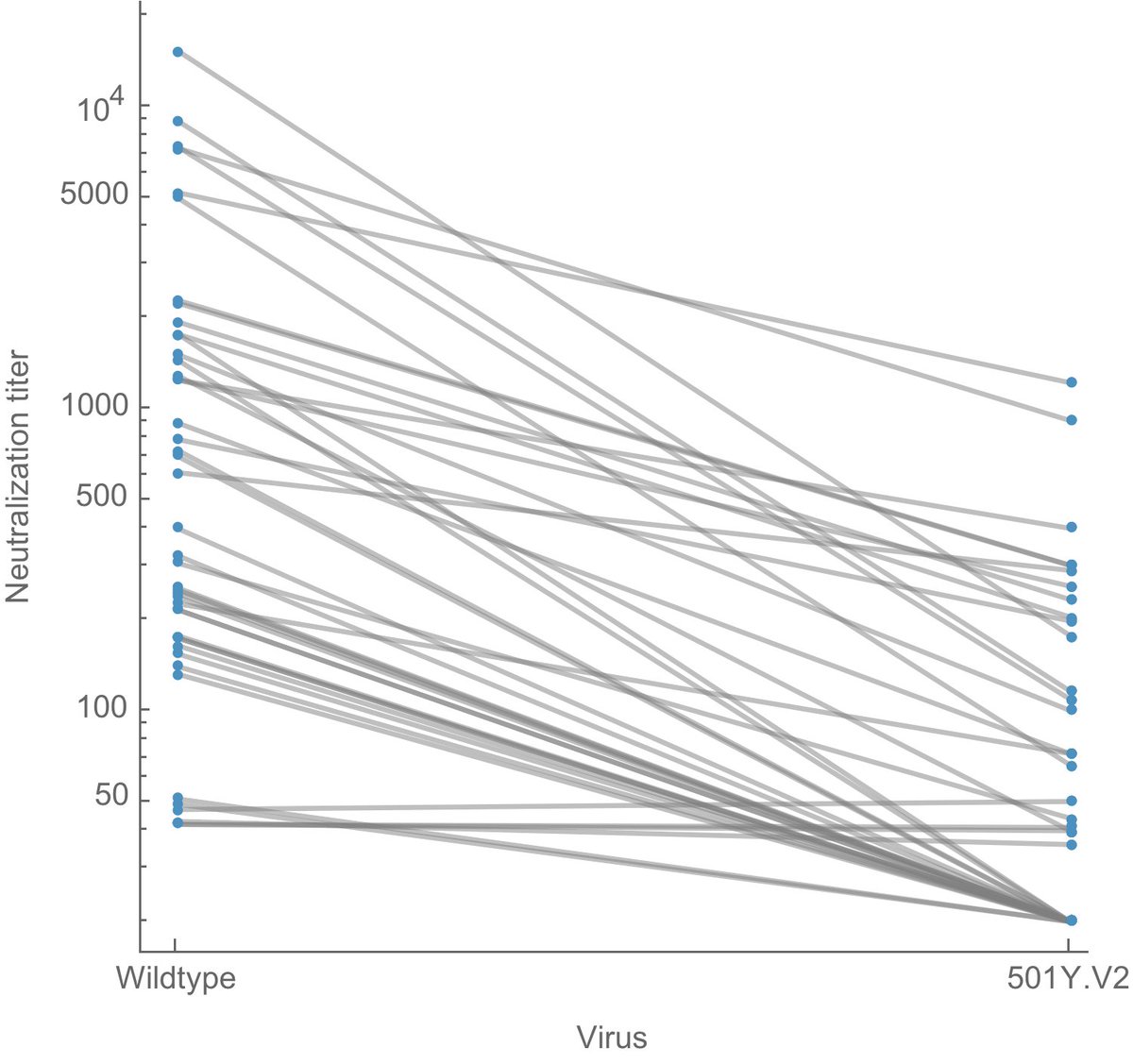
When variants of concern were first identified in late Dec, the US was not where it needed to be in terms of genomic surveillance. However, with considerable ramp up by the CDC, state labs and academic groups, we now have a remarkable genomic surveillance system. 1/14
My favorite metric for genomic surveillance is the number of cases that have been sampled, sequenced and shared publicly to @GISAID in the previous 30 days. By incorporating both sequencing volume and turnaround time, it tells you how much is known about current circulation. 2/14
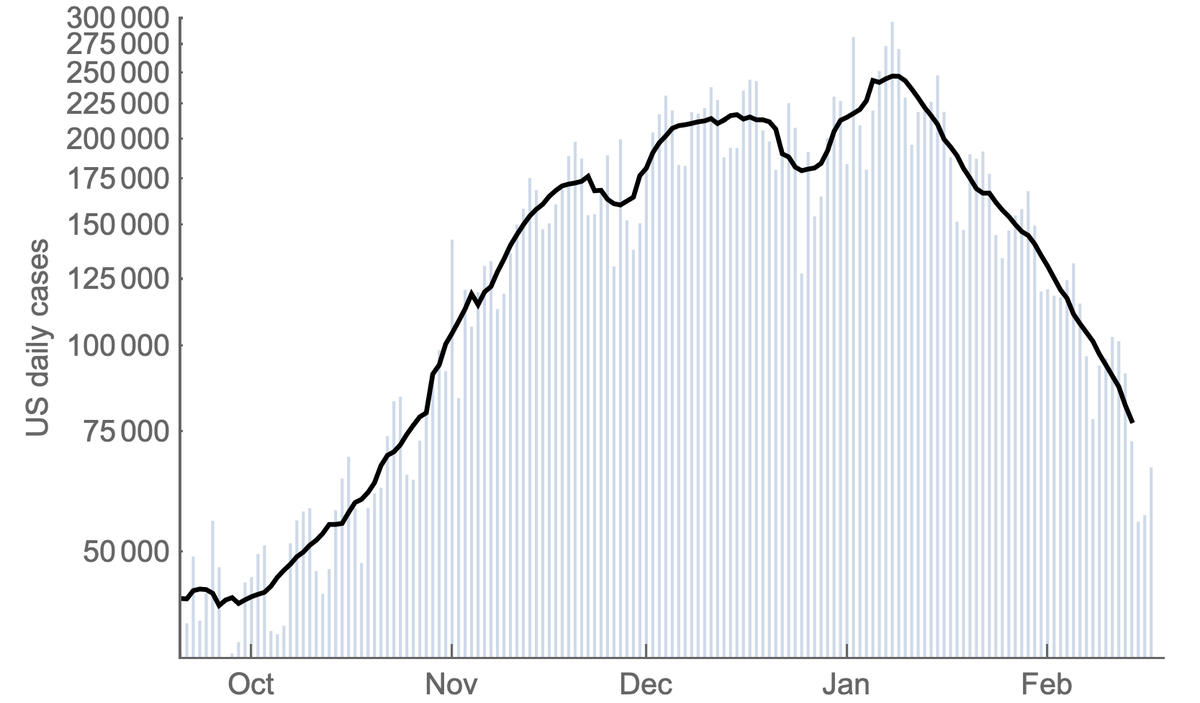
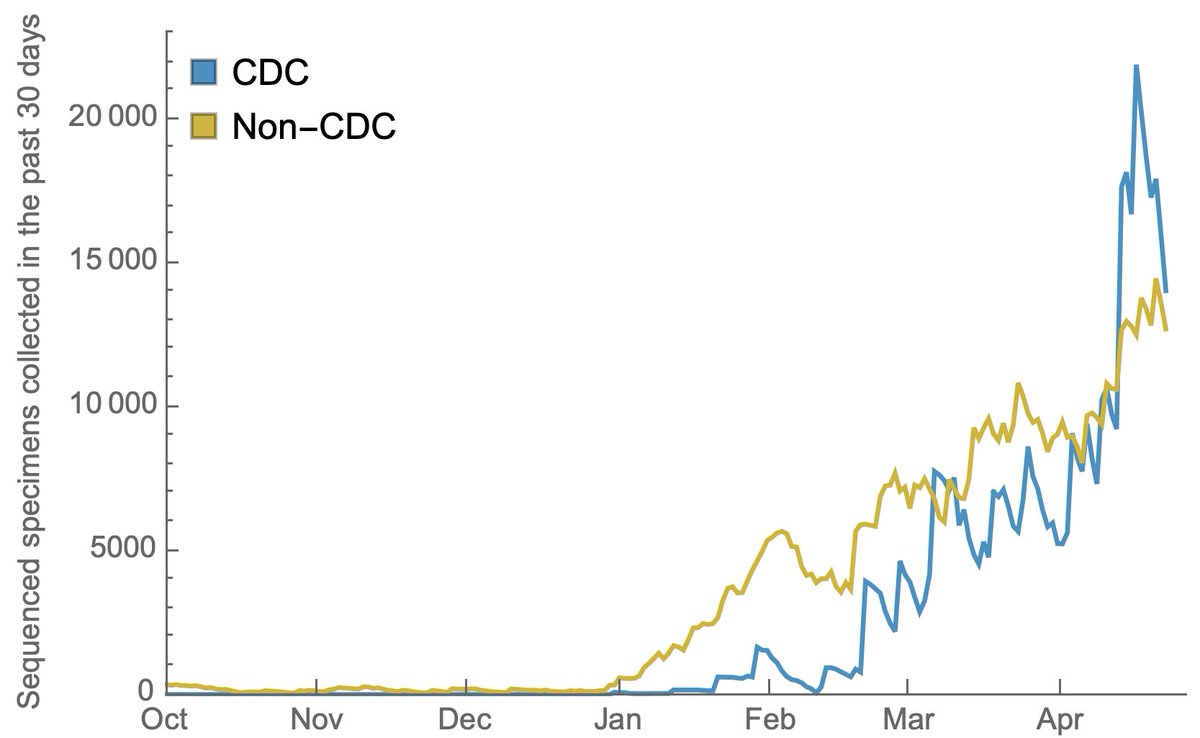
Throughout the fall, the US had just 100-300 genomes available that were sampled, sequenced and shared in the previous 30 days. 3/14
https://twitter.com/trvrb/status/1341806668045111296
However, with investments in sequencing capacity, the US had rapidly increased this count, ramping from 100-300 in Dec 2020 to over 30,000 in April 2021. This is a remarkable achievement and I'm no longer worried about novel variants escaping detection in the US. 4/14 
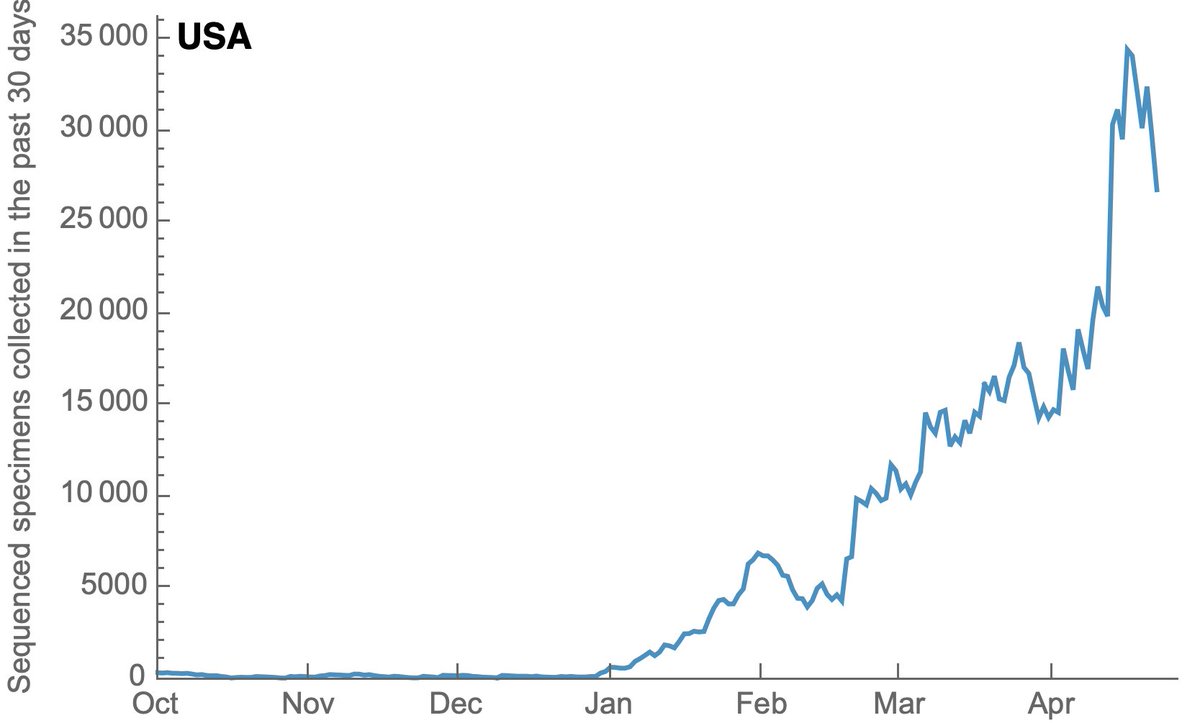
This increase in sequencing has been driven by the CDC and by non-CDC groups (mostly academic labs and state lab health departments). 5/14
Much attention has focused on proportion of cases sequenced, where in the past 30 days the US has sequenced about 1.7% of cases, behind countries such the UK (30%), Switzerland (5%) and Germany (2.5%). Data from gisaid.org/index.php?id=2…. 6/14
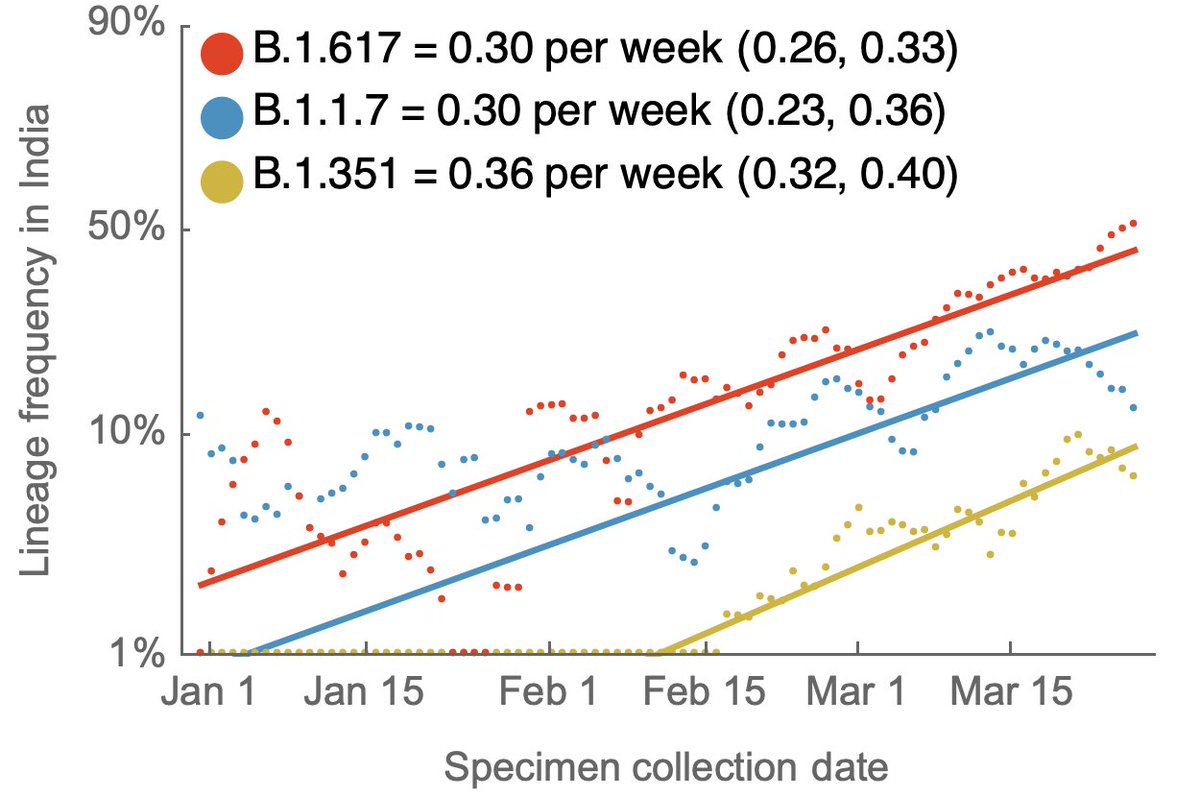
However, for variants in particular, analysis focuses on frequency, where we particularly care about variants that are increasing rapidly in frequency across different geographies. 7/14
In this case, having a very large volume of sequences with good turnaround time should be sufficient to characterize variants while they're still at low frequency. 8/14
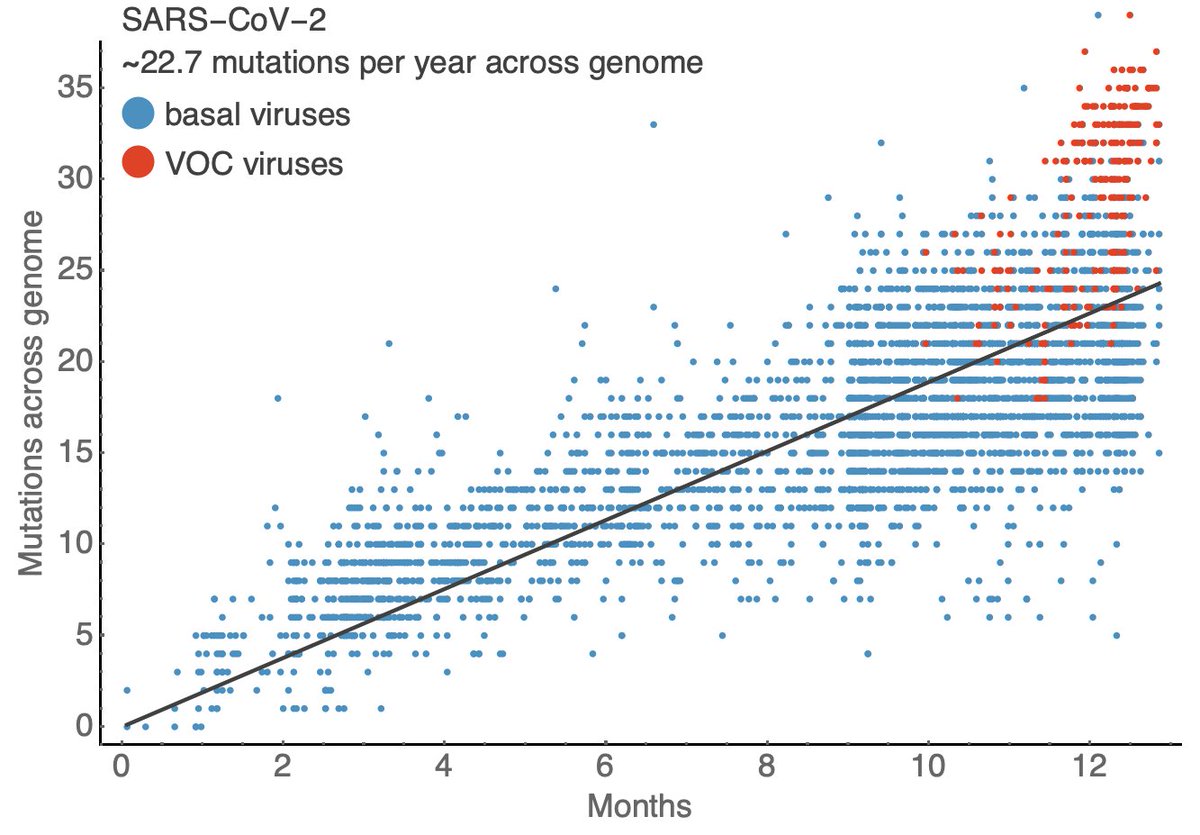
With ~30,000 genomes in the past 30 days, we can reliably catch variants at a 0.02% frequency threshold in the US (geometric distribution with 99% probability of detection). Again, this is remarkable. 9/14
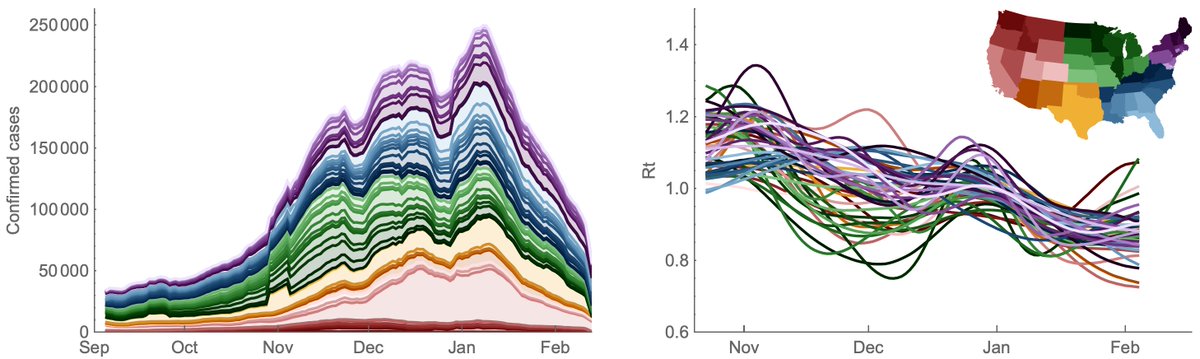
Other countries have also been increasing sequencing throughput in response to the emergence of variants of concern and we see the US now matching sequencing throughput of the UK and the rest of Europe when considered separately. 10/14
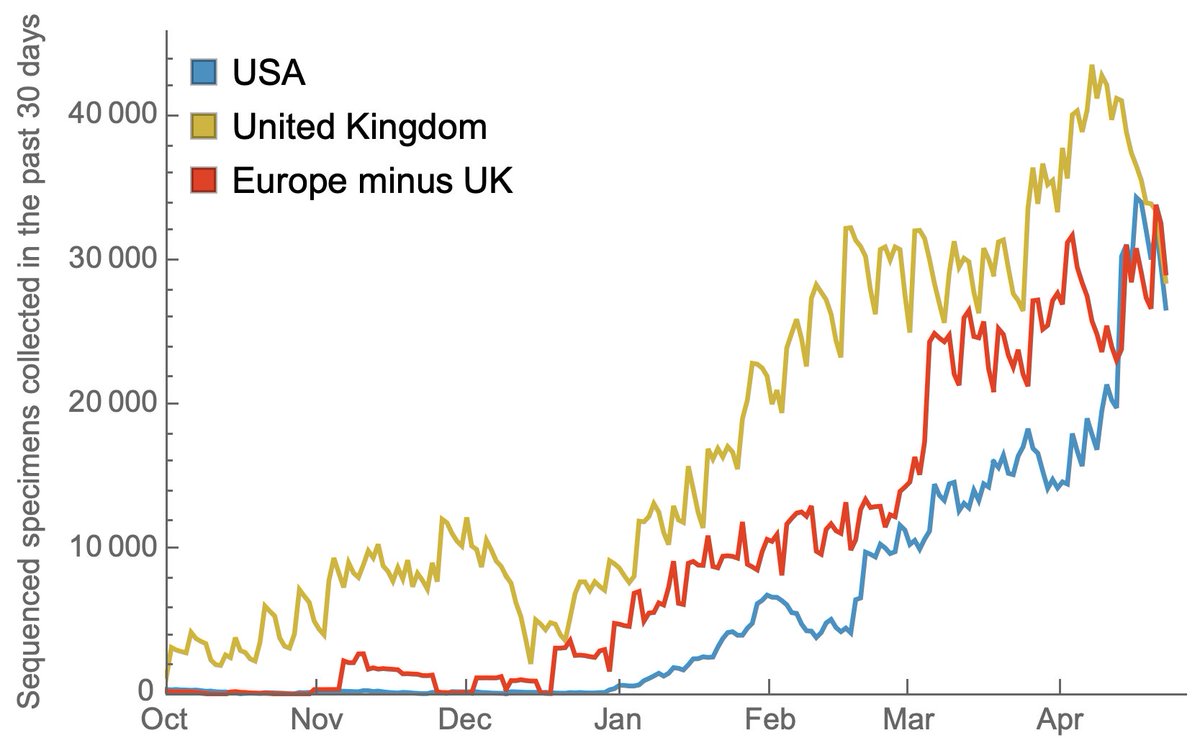
However, SARS-CoV-2 variants are emerging throughout the world and only focusing on improving genomic surveillance within national borders is short sighted. The primary goal of this surveillance is to be to able to formulate vaccine updates with sufficient lead time. 11/14
Work by South African and Brazilian scientists to quickly identify and share data on B.1.351 and P.1 did the world an enormous favor. 12/14
Sequencing throughput has been increasing in Africa and South America, but now lags behind the US and Europe due to recent national investments in sequencing capacity in the US and Europe. 13/14
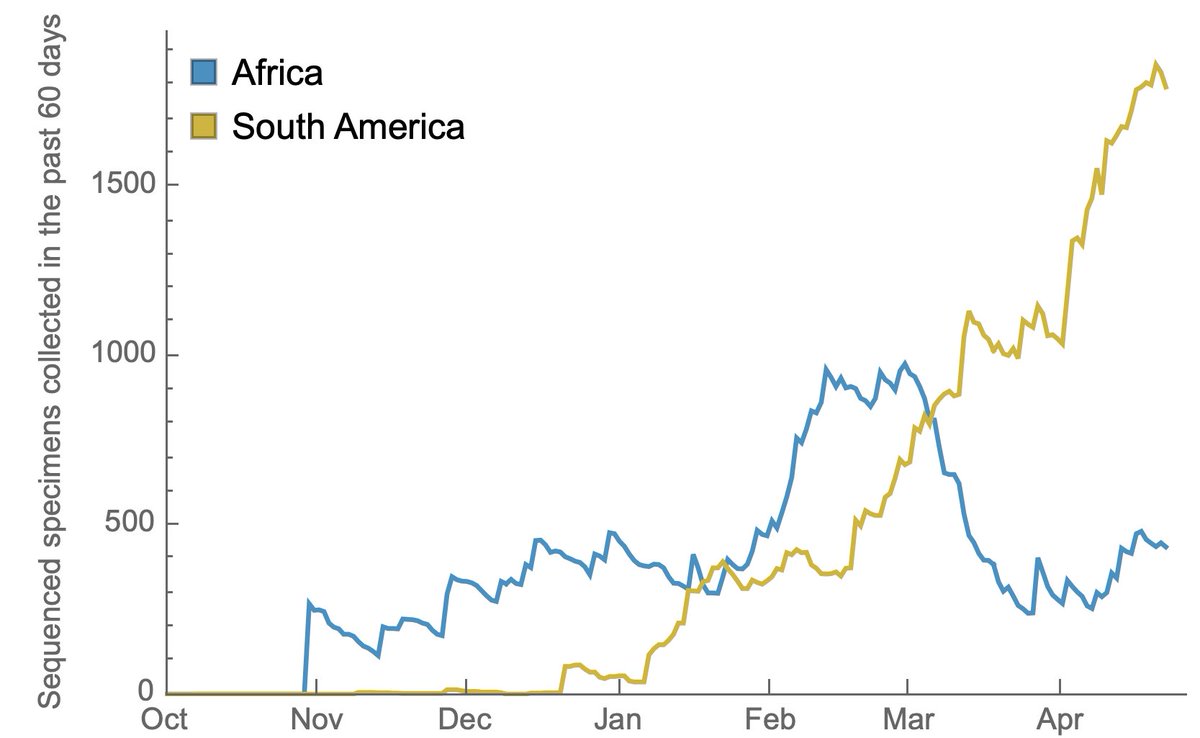
International investment is incredibly important here. I'd so much prefer an additional 1000 genomes from South America or Africa to an additional 10,000 from the US or Europe. 14/14
• • •
Missing some Tweet in this thread? You can try to
force a refresh