1/8 @DarrenM98230782 and I summarize our thoughts on the near-term evolutionary fate of the #Omicron lineage and its individual mutations
(virological.org/t/a-fool-s-err…). As the title implies, prediction of viral evolution has not historically been successful.
(virological.org/t/a-fool-s-err…). As the title implies, prediction of viral evolution has not historically been successful.
2/8 Anyone who claims to *know* where this is going is a fool. The emergence of #Omicron testifies to it. We consider four scenarios for the lineage in the near/midterm. This is also purely from an evolutionary perspective; the ultimate clinical outcomes are a separate story.
3/8
1. Spread with no selective pressure [unlikely]
2. Spread with weak/moderate ongoing selective pressures [most likely]
3. Spread with strong ongoing selective pressures [new data will soon show how likely]
4. Recombination wildcard [not too likely]
1. Spread with no selective pressure [unlikely]
2. Spread with weak/moderate ongoing selective pressures [most likely]
3. Spread with strong ongoing selective pressures [new data will soon show how likely]
4. Recombination wildcard [not too likely]
4/8 Scenario 2 is consistent with with a general selective sweep, where #Omicron becomes the dominant strain and evolves relatively slowly without significant changes in function/transmissibility/resistance. This would mirror the spread of Alpha and Delta.
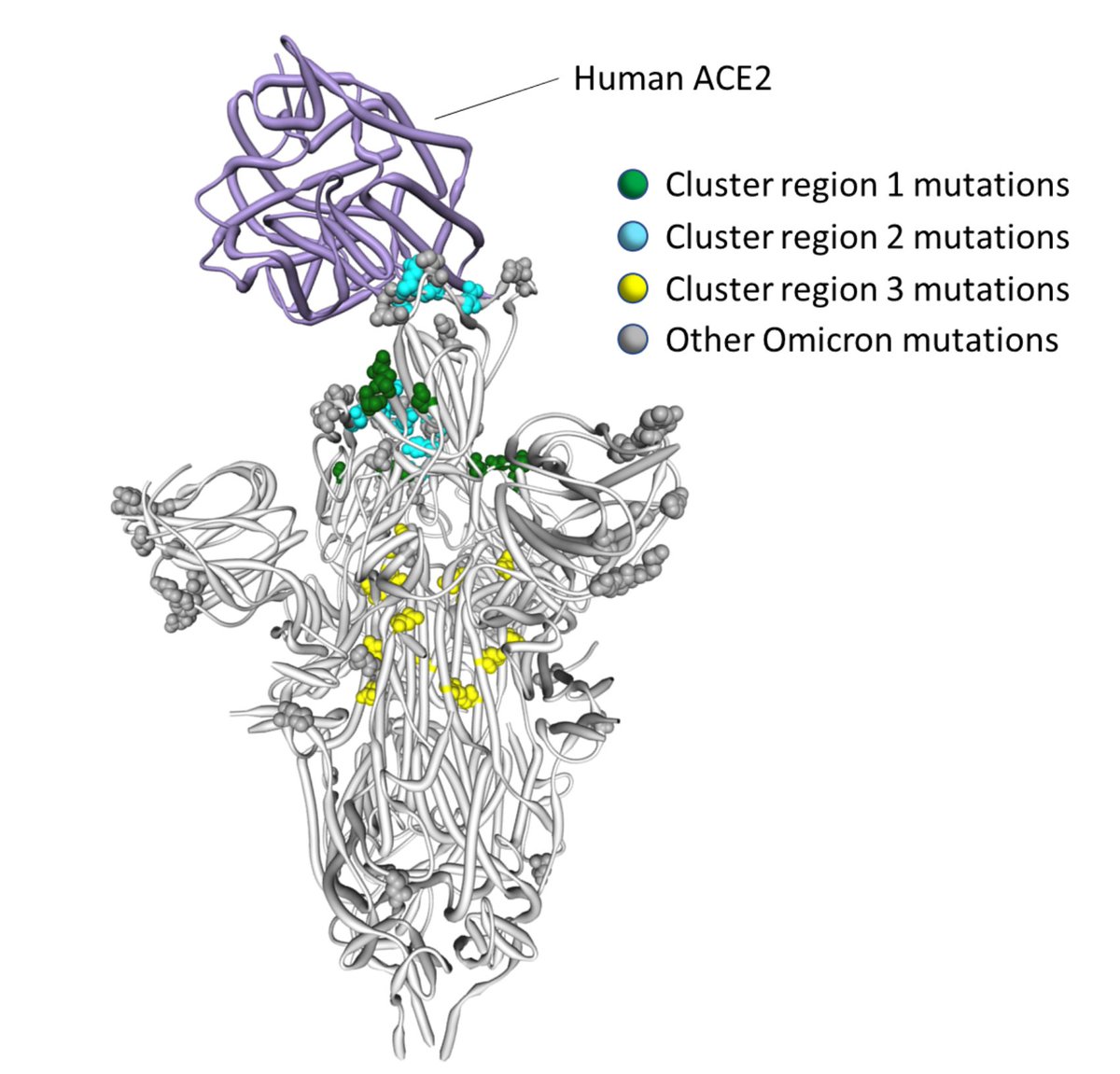
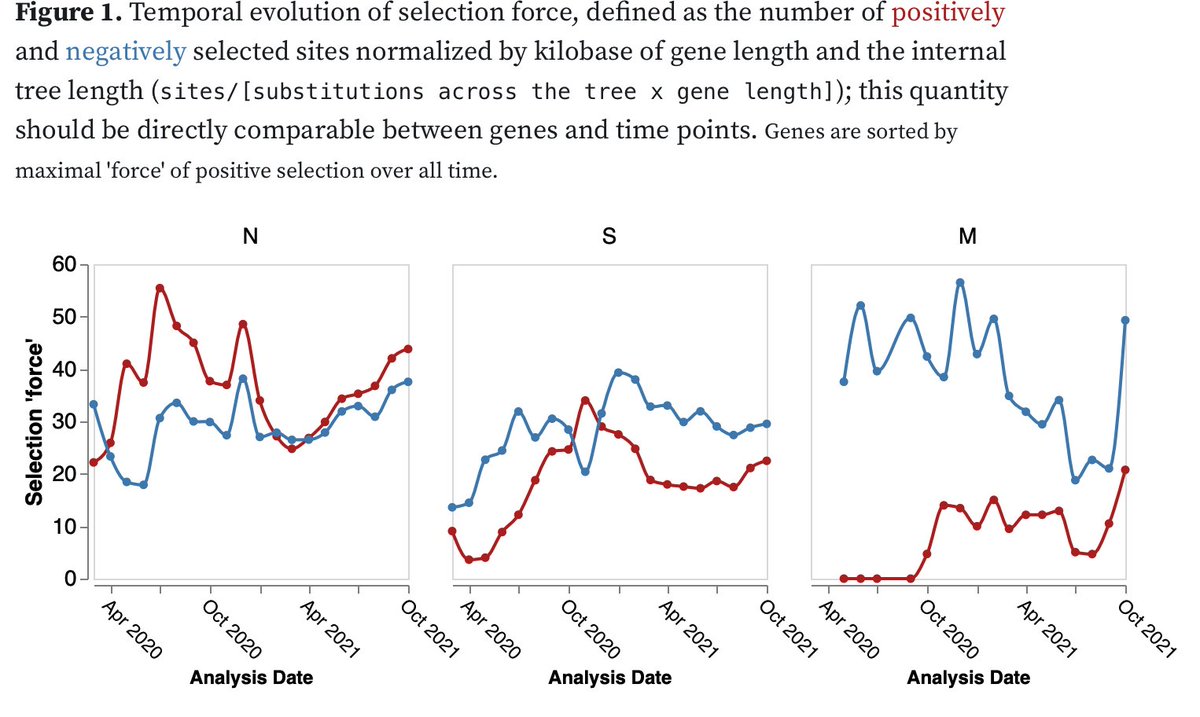
5/8 More interestingly, we expect there to be several detectable and interpretable patterns in evolution at specific sites in #Omicron. Our current monitoring and evolutionary analysis capacity can be used to better understand the evolutionary past and future of this lineage.
6/8
1. Population-level maladaptive mutations, if carried over from intra-host evolution, will revert.
2. Adaptive mutations at previously negatively selected sites should be maintained by negative selection.
1. Population-level maladaptive mutations, if carried over from intra-host evolution, will revert.
2. Adaptive mutations at previously negatively selected sites should be maintained by negative selection.
7/8
3. We expect some of the mutations seen in the 501Y meta-signature to appear in the Omicron lineage (e.g. 5F,18F, 701V etc) and rise in frequency over time
4. It is especially important that the acquisition of mutations that could confer additional immune escape is monitored.
3. We expect some of the mutations seen in the 501Y meta-signature to appear in the Omicron lineage (e.g. 5F,18F, 701V etc) and rise in frequency over time
4. It is especially important that the acquisition of mutations that could confer additional immune escape is monitored.
8/8
5. We would expect all the “easily accessible” large effect immune escape and transmission advantage mutations to arise quite rapidly.
6. We should make better use of deep sequencing data to investigate intra-host variability, especially occurring at sub-consensus levels.
5. We would expect all the “easily accessible” large effect immune escape and transmission advantage mutations to arise quite rapidly.
6. We should make better use of deep sequencing data to investigate intra-host variability, especially occurring at sub-consensus levels.
• • •
Missing some Tweet in this thread? You can try to
force a refresh