Hace un mes reportamos una nueva variante de SARS-CoV-2 que se expande rápidamente en Sudamérica. Le llamamos C.37, aunque algunos ya le llaman "variante andina". Les cuento lo que sabemos hasta hoy.
1/25
1/25

Empecemos diciendo que preferimos no llamar a las variantes SARS-CoV-2 por su país de orígen (británica, sudafricana, etc) ya que asume que la evolución de variantes es responsabilidad de estos países y alimenta nacionalismos innecesarios en respuesta a un desafío global.
2/
2/
Este update se basa en datos subidos a @GISAID. Podemos analizar secuencias genómicas de todo el mundo porque los países e investigadorxs que los generan los comparten de forma abierta y transparente. ¡Van 1,6 millones de genomas publicados!
3/
3/

Pero la mayoría de genomas viene de países ricos. Sudamérica ha generado 20,000 genomas, que son el 1.2% del total en GISAID y menos del 1% de casos en la región. Con tan poca vigilancia, nuestra capacidad de detección de variantes es muy limitada. bit.ly/3hDC0lP
4/
4/

Y los datos no sirven de mucho por sí solos: deben ser clasificados y analizados continuamente. Usamos @nextstrain y outbreak.info, dos excelentes herramientas gratuitas producto de colaboraciones multidisciplinarias e ideales para data nerds como yo. 🤓
5/
5/

Les dejo el enlace a outbreak.info para que puedan seguir las novedades sobre C.37 y sus variantes favoritas. Aquí les presento un resumen de su reporte.
6/
outbreak.info/situation-repo…
6/
outbreak.info/situation-repo…
C.37 presenta en su genoma 19 mutaciones no sinónimas (que generan un cambio en la secuencia de aminoácidos). Siete de éstas ocurren en la proteína Spike, principal antígeno del virus.
7/
7/

Aquí un detalle de las mutaciones de C.37 comparada con las variantes de preocupación (VOCs) B.1.1.7, B.1.351, P.1 y B.1.617.2 (la más reciente, asociada a la India) y B.1.1.1 (una variante regular que posiblemente dio origen a C.37)
8/
8/

Sabemos muy poco sobre el efecto de esta combinación de mutaciones sobre la biología del virus. Pero hay cuatro que (preliminarmente) preocupan: (1) deleción de 3aa en ORF1a, compartida con las VOCs B.1.1.7, B.1.351, P.1, y cuyo efecto se desconoce.
9/
9/

(2) deleción 247-253 en la región N-terminal de Spike; (3/4) mutaciones L452R y F490S en el motif de unión al receptor (RBM) de Spike.
10/
10/

Al 20 de mayo, se han reportado en GISAID 823 genomas de C.37 en 18 países, incluyendo Chile (n=276), Argentina (53), Peru (52), EEUU (337) y Alemania (53).
11/
11/

A partir de estos datos se pueden hacer estimaciones generales de prevalencia en cada país. Estas son sesgadas por varios factores y probablemente son un subreporte de la prevalencia real. Pero sugieren que C.37 circula principalmente en Sudamérica.
12/
outbreak.info/situation-repo…
12/
outbreak.info/situation-repo…


En Perú, no tenemos información nueva desde abril. Nuestro lab en UPCH recién recibió los fondos de Concytec esta semana (10 semanas después de ganar el concurso) y estamos a la espera de reactivos para generar nuevas secuencias.
13/
13/
Hace unos días se anunció que INS recibirá fondos adicionales para generar 1,200 secuencias *mensuales* en 2021. Es una gran noticia pero deja de lado a las universidades. Pronto compartirán sus nuevas secuencias en GISAID y sabremos la prevalencia actual de C.37 en Perú.
14/
14/
El Presidente visitó INS la semana pasada para supervisar el escalamiento de la vigilancia genómica. No le dijeron que el secuenciador mencionado (NextSeq 550) ya existe en UPCH y UNTRM (Amazonas) desde hace más de dos años. En fin...
15/
15/

Chile tiene un mejor sistema de vigilancia y ya lleva 998 genomas publicados en 2021. 276 (28%) son C.37, 247 (25%) son P.1 y 70 (7%) son B.1.1.7. Pero da la impresión que aún no reconocen la existencia de C.37.
bit.ly/3vjEeKU
16/
bit.ly/3vjEeKU
16/


En Argentina, @ProyectoPAIS4 reporta desde febrero un aumento de la mutación L452R presente en C.37. En abril, C.37 fue el 33% de los genomas de casos no asociados a viajes en Buenos Aires, compitiendo con P.1 y B.1.1.7.
bit.ly/3f5IsAr
17/
bit.ly/3f5IsAr
17/

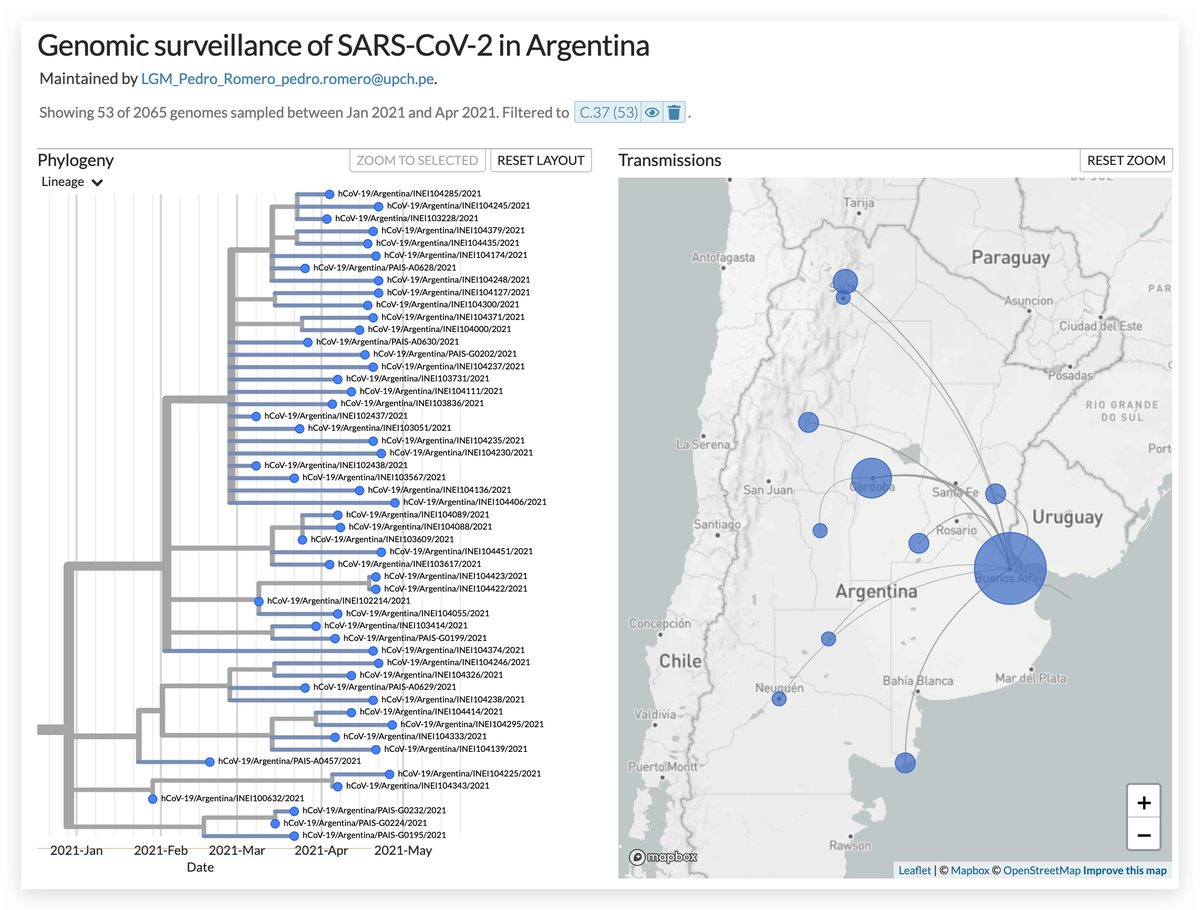
Aquí otra forma de visualizar los genomas de Argentina, via Nextstrain. Elaborado por @quipupe con datos de GISAID (n=2,065) @JJocampos23 @Poklepovich
18/
18/

También se reportado recientemente en Uruguay. @goyoiraola
19/
19/
https://twitter.com/Dr_Julio_Medina/status/1391161772581732352
Llama la atención EEUU, que reporta 337 genomas de C.37 en 30 estados, con clara relación a los casos de Sudamérica. EEUU secuencia miles de genomas por semana, por lo que su prevalencia sigue siendo baja. Pero se nota una tendencia al aumento.
21/
21/



No sabemos aún si es más transmisible o más letal. Para eso se requiere MUCHA información que aún no tenemos. Aquí @teozka les explica cómo se estima la mayor transmisibilidad de B.1.617.2 en UK y la evidencia que soporta su clasificación como VOC.
22/
22/
https://twitter.com/teozka/status/1394920382478618625
¿Sirven las vacunas contra C.37? Para eso también necesitamos datos adicionales basados en infecciones in vitro, modelos animales, ensayos de neutralización, etc. Pero debemos recordar que, hoy, todas las vacunas protegen contra las variantes conocidas
23/
23/
https://twitter.com/juanmoreb/status/1395049814522531843
Es imposible predecir la evolución de las variantes y su efecto sobre el curso de la pandemia. Pero podemos mantener la vigilancia genómica y observar su evolución en tiempo real. Nos corresponde informar a otros países sobre las variantes que tenemos en la región.
24/
24/
Eso es todo lo que tenemos por ahora. ¡Gracias por leer! Seguiremos informando.
Y no se olviden que #SinCienciaNoHayFuturo ✊
25/25
Y no se olviden que #SinCienciaNoHayFuturo ✊
25/25

Me adelanté en mi comentario al leer sobre el tema del reporte de C37 en Israel. De todas maneras, ilustra bien el problema de asociar variantes a países. No queremos líos con Israel.
27/
lanacion.cl/israel-descart…
27/
lanacion.cl/israel-descart…
Pero las secuencias son reales y ayer reportaron 23 C.37 de casos en mayo, posiblemente relacionados a una sola introducción a Israel desde América.
28/
28/


Corrección #2: Argentina secuencia una parte del genoma, el gen S, para optimizar la vigilancia y luego confirma algunos con genoma completo (lo entendí bien?). Gracias, @humbertodebat. Mi figura (modificada del reporte de PAIS) debió decir:
29/
29/

Vale mencionar que L452Q está asociada casi exclusivamente a C.37, aunque hay otros casos reportados en la región en los que L452Q está presente en otros 'backgrounds'
30/
30/

Un abrazo a lxs colegas de Chile + Argentina y gracias por los comentarios ✌️
31/
31/
• • •
Missing some Tweet in this thread? You can try to
force a refresh