
first, let me say it's been a super fun project and a huge tour-de-force from our very talented @CrowellHL .. and lucky for us, we benefit from the ever-present wisdom of @CSoneson, some great insights from Pierre-Luc Germain along the way and great collaborators at Roche.
So .. people talk a lot about differential expression in scRNA-seq data, but from what I can tell, most of it is about how to find marker genes. That's indeed interesting, but not what we are on about here. Here, we're interested in *sample-level* comparisons of scRNA-seq ..
meaning, we have a proper replicated experiment, multi-sample, multi-condition and we are profiling multiple cell subpopulations (e.g., tissue, blood, etc.). Here, we're interested in following a subpopulation (e.g., T cells) across samples, say to compare treated vs control.
We call this *differential state* analysis. The cells are already organized into "types" (ok, this requires a computational analysis) and we look for differences in expression in the cells of treated samples versus cells of control samples; we do this one subpopulation at a time.
Turns out there are many ways to do this: mixed models, "pseudobulks", comparing distributions. So, we wanted to survey how to best do this.
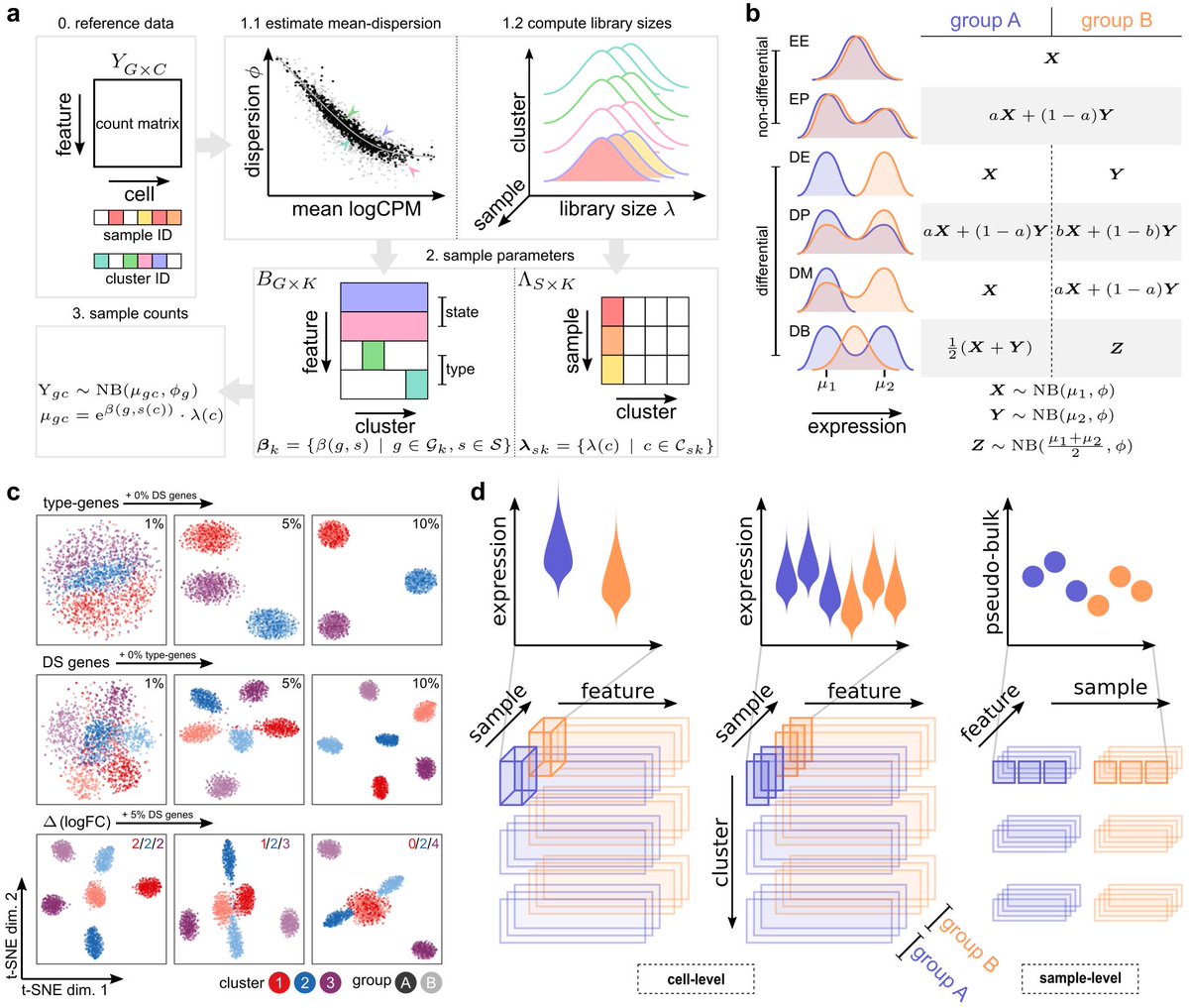
First hurdle was simulating data in a way that we maintain properties of scRNA-seq but also considers sample-to-sample variability.
First hurdle was simulating data in a way that we maintain properties of scRNA-seq but also considers sample-to-sample variability.
Not easy, but after some iterations (we test it against real datasets using @CSoneson's countsimQC package + some pseudobulk-level dispersion-mean plots to check sample-sample variability), I think we have a very flexible simulation .. 
After that, we do the bakeoff. Many more details in the paper, but overall pseudobulks do quite well and are fast. Mixed models work well, but bit of an extra cost to compute. Overall, I think there is scope for new methods too, especially if there is concern about aggregating.

We also did a nice analysis of an snRNA-seq dataset from mouse cortex tissue .. replicates of mice +/- peripheral LPS treatment .. and also, full code for the pipeline: integration, clustering, annotation, differential state analysis, subpopulation-level geneset analysis and more
Software: bit.ly/2YoCnax
Code: bit.ly/2Mj9mXB
Feedback welcome!
I will speak about this at #JSM2019 next week and we have a workshop on the topic @BC2Conference ..
Code: bit.ly/2Mj9mXB
Feedback welcome!
I will speak about this at #JSM2019 next week and we have a workshop on the topic @BC2Conference ..









