
A Thread (🧵)
Let's discuss long-read #sequencing, optical mapping, and the implications of a recent study (linked below).
Please view my disclosures at the end. I've intentionally made this thread more technical as I think it's necessary.
biorxiv.org/content/10.110…
Let's discuss long-read #sequencing, optical mapping, and the implications of a recent study (linked below).
Please view my disclosures at the end. I've intentionally made this thread more technical as I think it's necessary.
biorxiv.org/content/10.110…
First, let's deconstruct the paper's title: "De Novo Assembly of 64 Haplotype-Resolved Human Genomes of Diverse Ancestry and Integrated Analysis of Structural Variation".
De novo (Latin: "Of New") assembly involves sequencing a genome without the help of a reference.
De novo (Latin: "Of New") assembly involves sequencing a genome without the help of a reference.
Assembling a #genome de novo is like solving a jigsaw puzzle without using the picture on the front of the box. You could start with the corners, assemble the edges, and try to fill in the rest using color- or shape-matching methods.
Let me use an analogy.
Let me use an analogy.
Imagine the jigsaw pieces are sequence reads, the heuristics you use to put the pieces together are assembly algorithms, and the final product (hopefully) is a complete reference genome, as shown in the drawing below. I'll extend this farther. 
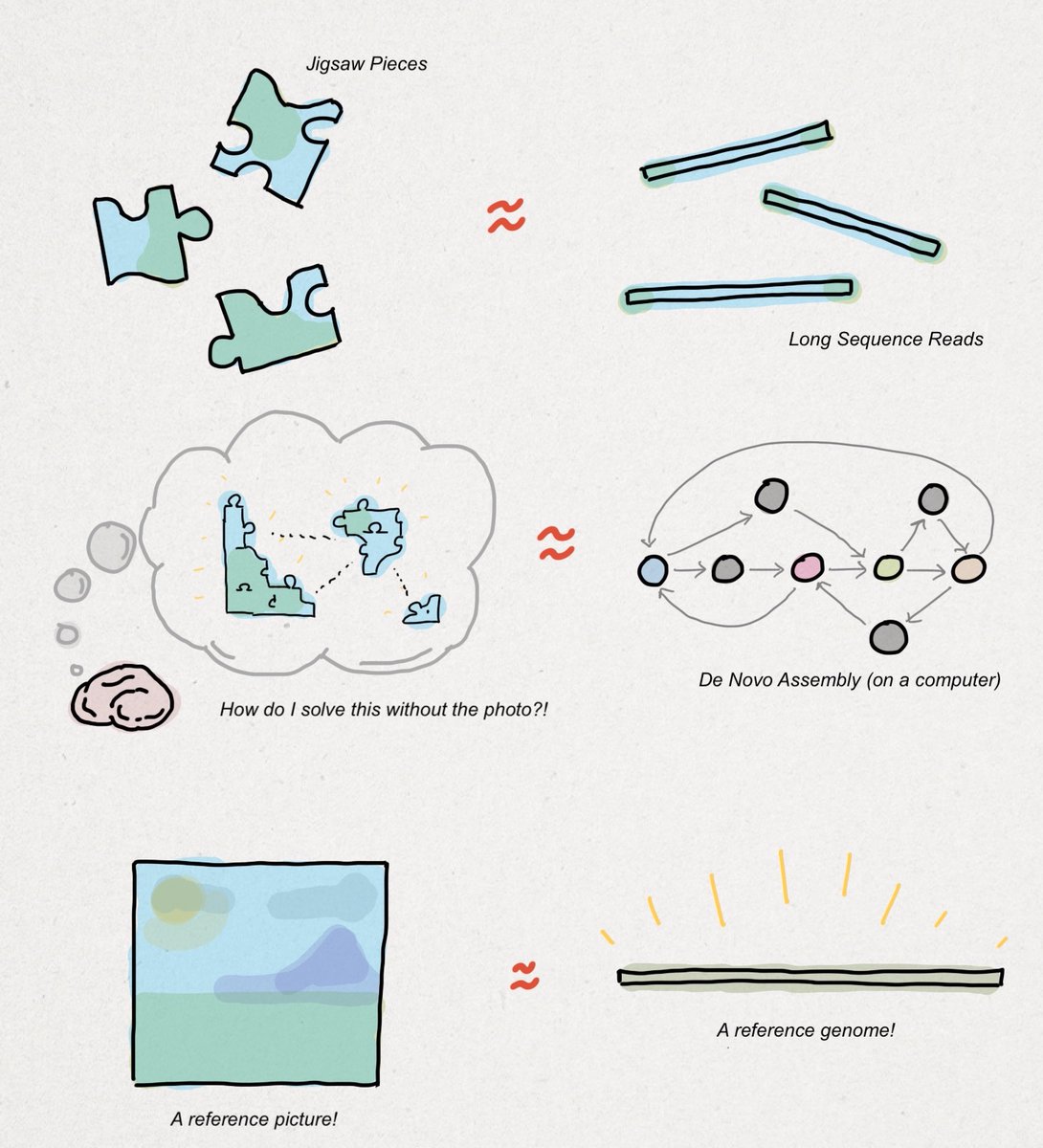
Most sequencing that goes on in the world is done with a reference just as most jigsaw puzzles are finished with the box nearby. That is to say, de novo sequencing is a substantially smaller market opportunity than #clinical sequencing.
Clinical sequencing works by comparing a patient's genome up to a reference genome. Intuitively, the differences are called #variants, as shown below. Detecting variants using a reference genome is easier the same way that solving a jigsaw using a photo is easier.
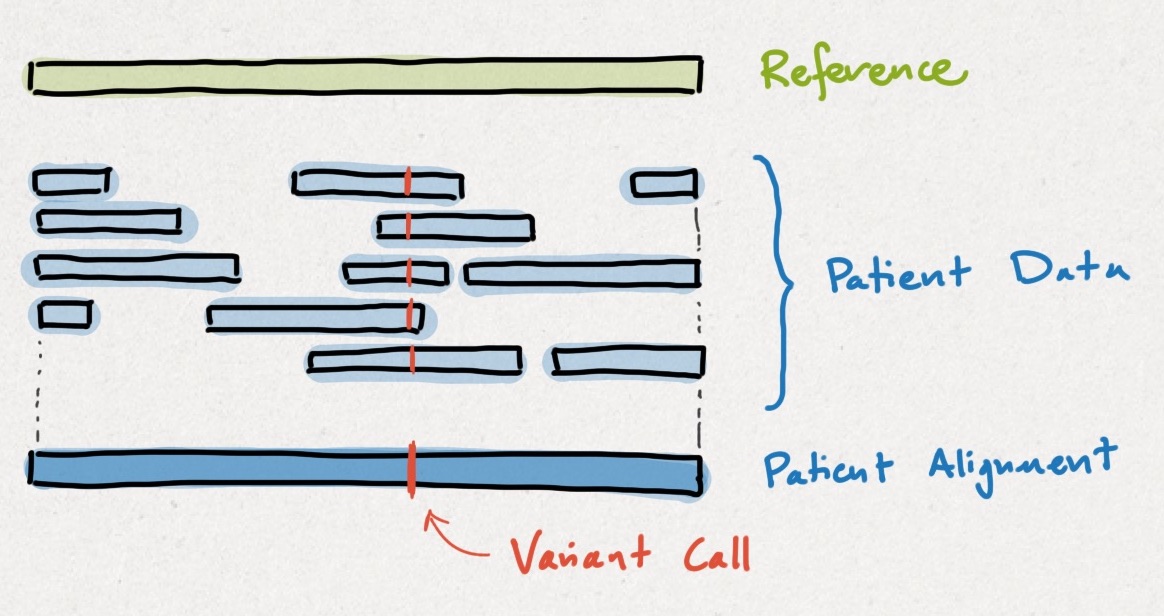
My point here, which I will reinforce later, is that conclusions drawn from this paper are most applicable to de novo sequencing, not clinical sequencing. This distinction will resurface again, so keep the jigsaw puzzle analogy in your back pocket.
What is a #haplotype-resolved genome?
Humans are #diploid organisms, meaning we have two copies of each gene - one from each parent. Each parental copy is a haplotype. When a variant is haplotype-resolved, that means we know which parent it was inherited from.
Humans are #diploid organisms, meaning we have two copies of each gene - one from each parent. Each parental copy is a haplotype. When a variant is haplotype-resolved, that means we know which parent it was inherited from.
The process of isolating variants to each parental haplotype is called phasing. Unlike a 'squashed' short-read assembly, a haplotype-resolved, long-read assembly tells researchers more about the functional consequences of genetic variants (also called annotation).
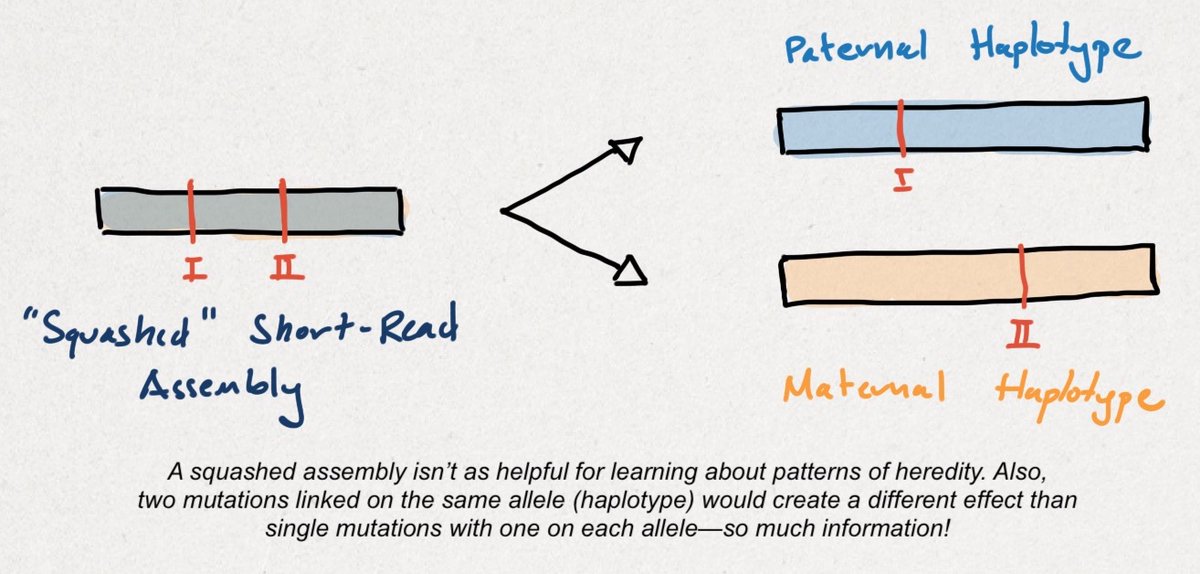
Importantly, the researchers' method was focused on an ethnically-diverse group of persons. By generating more diverse reference genomes, we can improve variant calling performance for everyone. A dangerous variant in one person could be benign for another!
Let's shift gears.
Let's shift gears.
I'm now going to discuss several methods and instruments, some of which were used in this paper. As a reminder, methods and instruments are NOT companies. Please don't misinterpret my opinions/critiques of technologies/tools as my feelings towards a company.
Similar to other de novo approaches, the researchers' method combined multiple sequencing approaches. I've outlined them below. This is common in de novo research settings where discovery rates are optimized over cost, turnaround time, or ease of use.
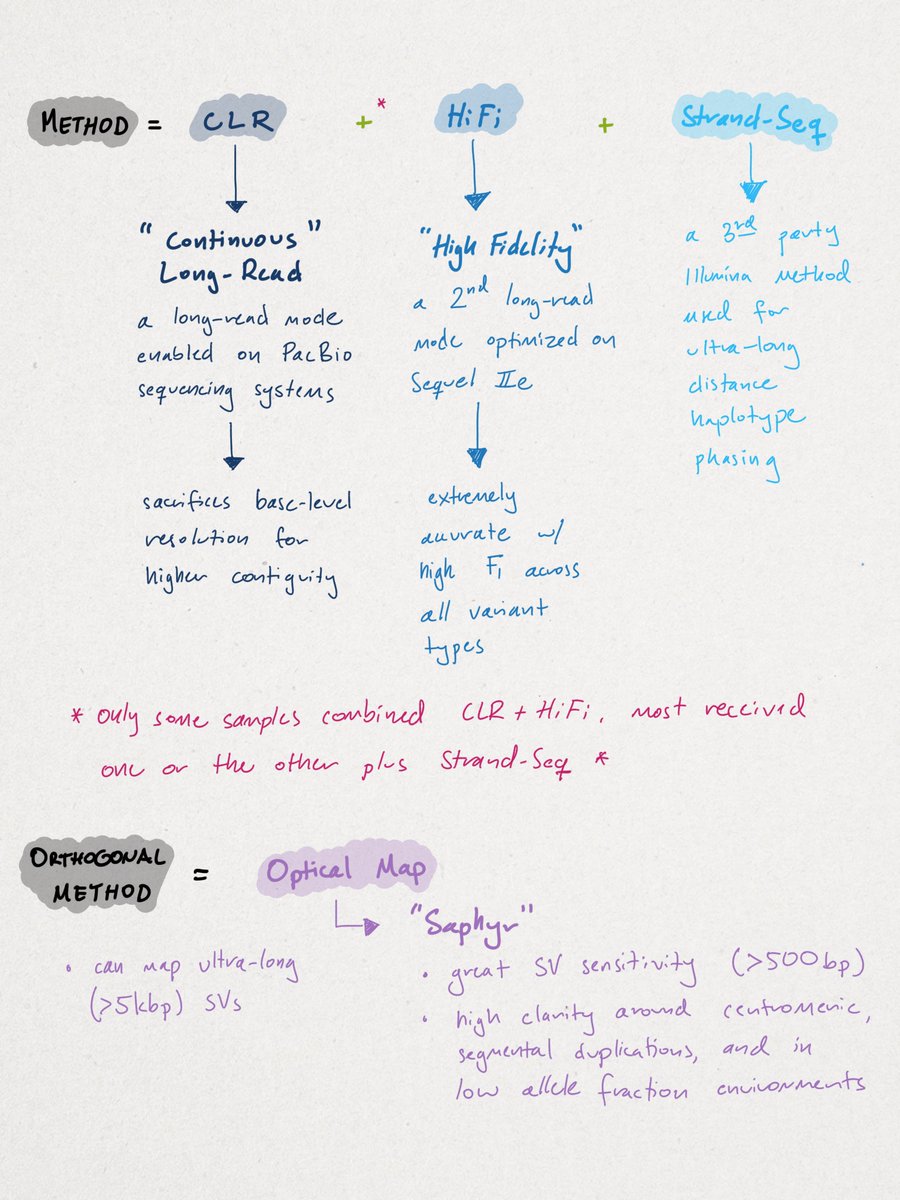
While this 'kitchen-sink' approach may apply to de novo sequencing, rarely have I ever seen a clinical setting wherein genomic data is fed from one sequencer to another. This is another reason why I think it's improper to generalize cost-comparisons derived from this paper.
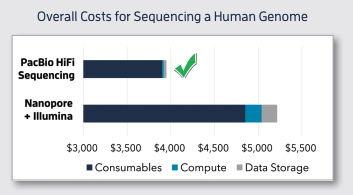
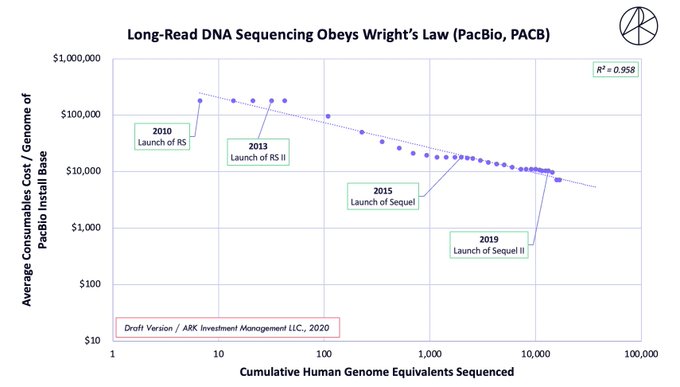
The pooling of methods may be why some believe LRS to be 20-40X more expensive (generally) than optical mapping, which is incorrect. For example, #WGS on the Sequel IIe is quoted at ~$3.7K, not $10-20K, which is how it would be used in a clinical setting (see cost-decline below).
Let's now discuss the study's results.
Across all samples, the researchers yielded 18,207,906 unique variants that break down in the following manner (see illustration below). We'll begin with the smallest variants (SNVs) and end with the largest structural variants (SVs).
Across all samples, the researchers yielded 18,207,906 unique variants that break down in the following manner (see illustration below). We'll begin with the smallest variants (SNVs) and end with the largest structural variants (SVs).
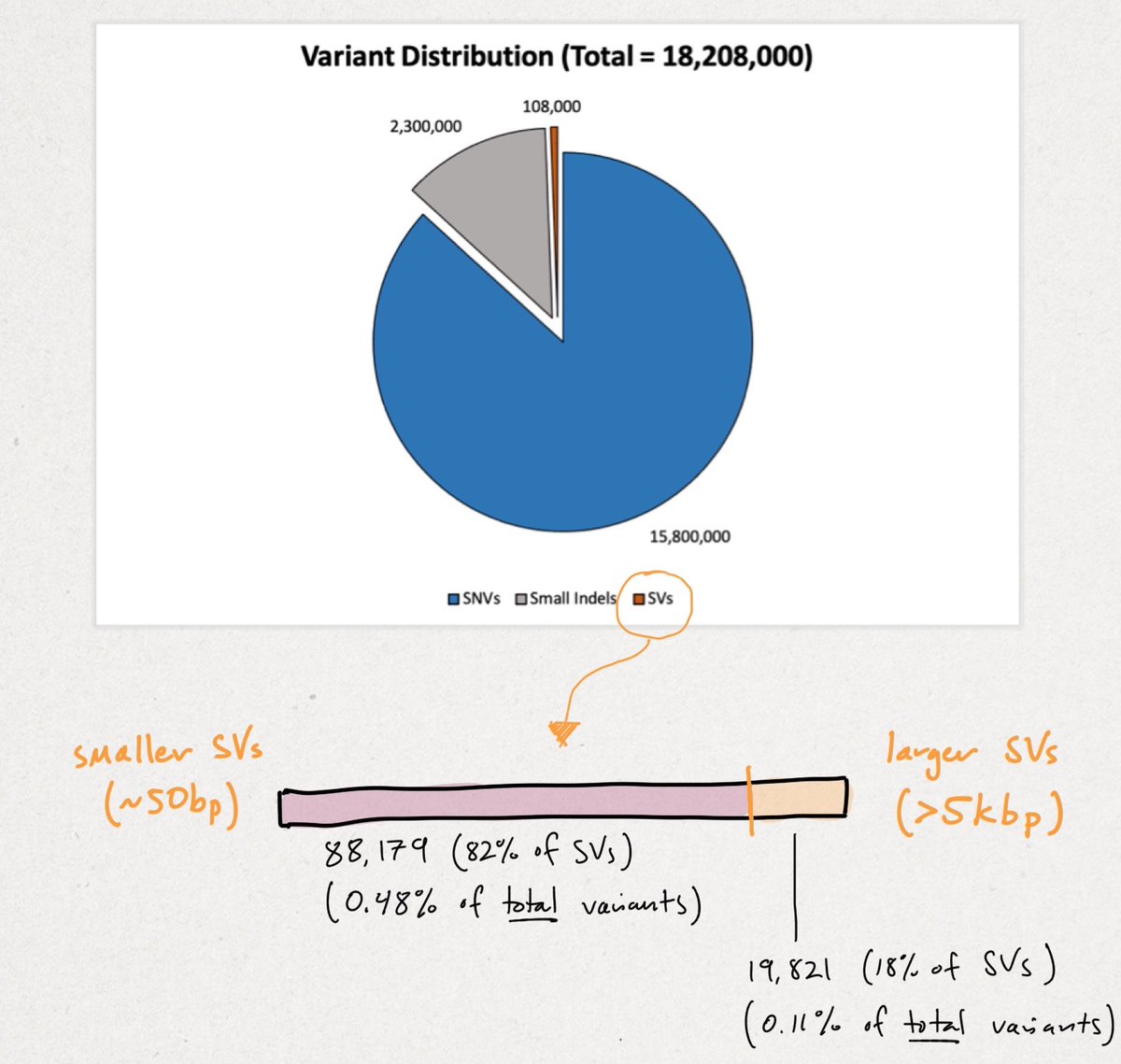
Recall there are ~3B base pairs (bps) in the human genome. SNVs are single letter (1 bp) change. An indel (portmanteau of insertion + deletion) is <49 bps. SVs vary widely in size, but begin at >50 bps up to tens-of-thousands of bps in size.
You can see the vast majority of variation is small. Though rarer SVs often impart larger functional changes. SVs tend to be highly repetitive (AAA) and/or contain repetitive motifs (ATGATGATG). This causes short-reads to fail, as referenced multiple times in the print.
HiFi, CLR, sequencing by synthesis (SBS), and #nanopore sequencing all aim to detect the maximum # of variants, though with varying degrees of success. I won't compare them here except to say we think short-reads are ineffective, even w/ improved graph-based assemblers.
As shown here, the researchers' method fully resolved nearly all variants, except for 28% of large SVs detected via OM. Though, if you consider partially-detected calls, OM adds only 6% more clarity. In fairness, I'll stick with the 28% because it doesn't change my point.
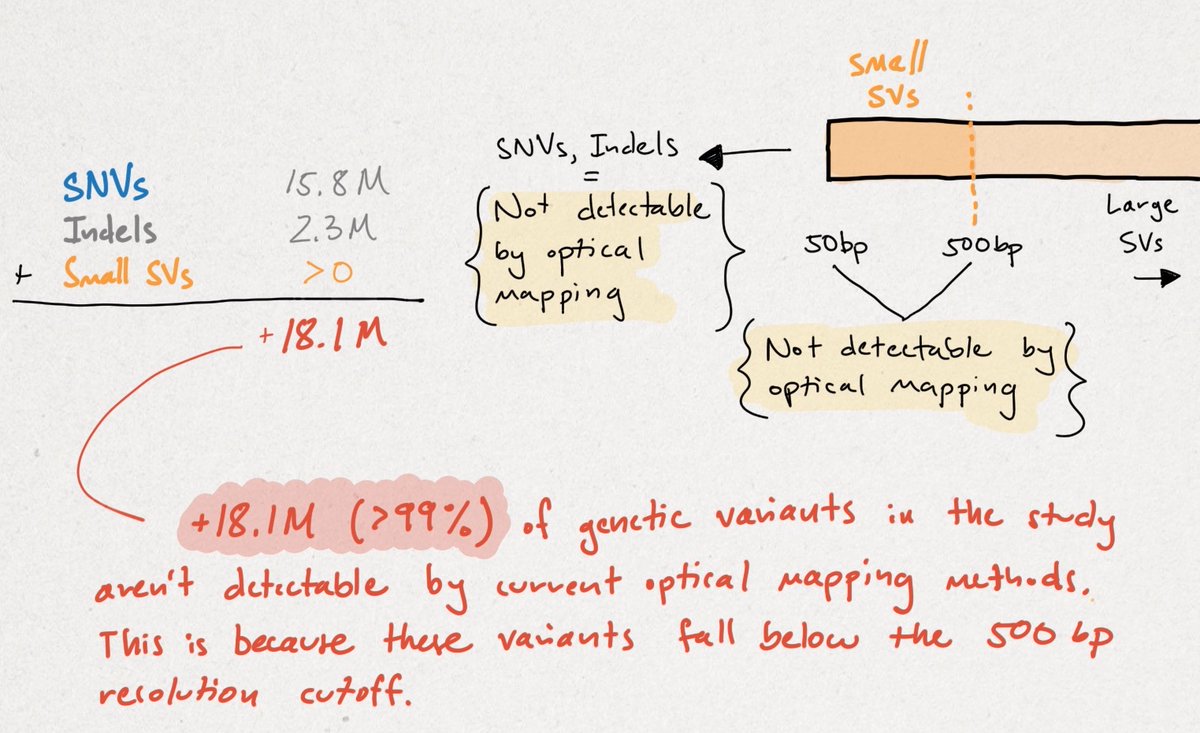
The conclusion should be that the HiFi/CLR + SS method identified >99% of the variation in this study, with OM adding an additional 28% of 1%, or objectively 0.028% more variant calls. OM isn't capable of calling variants <500 bp, which is 99% of the total variation.

This does NOT mean that OM isn't important, needed, or exciting. I think it's all three, but to say that LRS detects only 72% of SVs as opposed to OM is incorrect. To say that LRS detects just 72% of LARGE SVs is more accurate, but omits where LRS technology is going.
We disagree outright that sequence reads aren't getting longer as it's clearly an R&D goal for both fluorescence and nanopore-based LRS providers. I'm unaware of the theoretical 'upper limit' for read-lengths, especially for nanopore-based methods.
In fact, the recent telomere-to-telomere (T2T) assembly of the most complete reference genome only used HiFi & ultra-long nanopore reads for long-range scaffolding, as shown below. I'd argue sufficiently longer reads will improve large SV calling.
genomeinformatics.github.io/CHM13v1/
genomeinformatics.github.io/CHM13v1/

Still, I agree in principle that massively complex SVs, segmental duplications (SDs), regions near centromeres, and very heterogeneous cancer samples w/ low #allele fraction are perfectly cut out for OM-first approaches.
Indeed, I'm equally excited for upcoming presentations on these areas in a few weeks. I agree, in principle, that OM will likely always outperform LRS for detecting the most esoteric SV events, though that margin may erode with time.
I've even written about some of these cases, such as chromothripsis, where I now believe OM has a key role to play in discovery. This print also shows how OM is vital to generating haplotype-resolved, de-novo genomes.
https://twitter.com/sbarnettARK/status/1234856636956762121
I'll admit another place where my knowledge is opaque. What does multiplexing look like on Saphyr? Throughput is one thing, which again I'll admit has improved a shocking amount, but can a lab do targeted work using UMI's for sample batching?
In conclusion, there's no single measure for 'better' in genomics. That's like saying a racecar is better than a Jeep Wrangler. It all depends on how fast you need to go and what rocks you may need to climb.
Disclosure(s):
General: bit.do/eyRo8
I'm focused on technology, products, and methods, not comparing companies. This is not a buy, sell, or hold recommendation for any security.
General: bit.do/eyRo8
I'm focused on technology, products, and methods, not comparing companies. This is not a buy, sell, or hold recommendation for any security.
Disclosure(s) Cont.
To YouTubers/vloggers, please do not spread rumors. Instead, try to hear out my angle, come up with your own take, or give insight where I may be wrong so I can learn.
To YouTubers/vloggers, please do not spread rumors. Instead, try to hear out my angle, come up with your own take, or give insight where I may be wrong so I can learn.
Edit: Should be 0.28%, not 0.028% more large SV calls.
• • •
Missing some Tweet in this thread? You can try to
force a refresh








