,
33 tweets,
13 min read
Read on Twitter
1/Newly revised on @biorxivpreprint: @MKarimzade's and my manuscript on Virtual ChIP-seq, our method for predicting transcription factor binding sites better through applying underutilized kinds of data doi.org/10.1101/168419
2/People have used position weight matrix (PWM) methods for predicting the binding of transcription factors since the 1970s. Unfortunately, in vivo, most of these predictions are useless, as stated by the futility conjecture of @WyWyWa and @albin_san nature.com/articles/nrg13…
3/Data on in vivo locations of transcription factors and other chromatin-associated factors from @ENCODE_NIH reveals the opposite problem—most of the locations found by biochemical assays like ChIP-seq don't have good PWM motif matches. We call this the dual futility conjecture. 
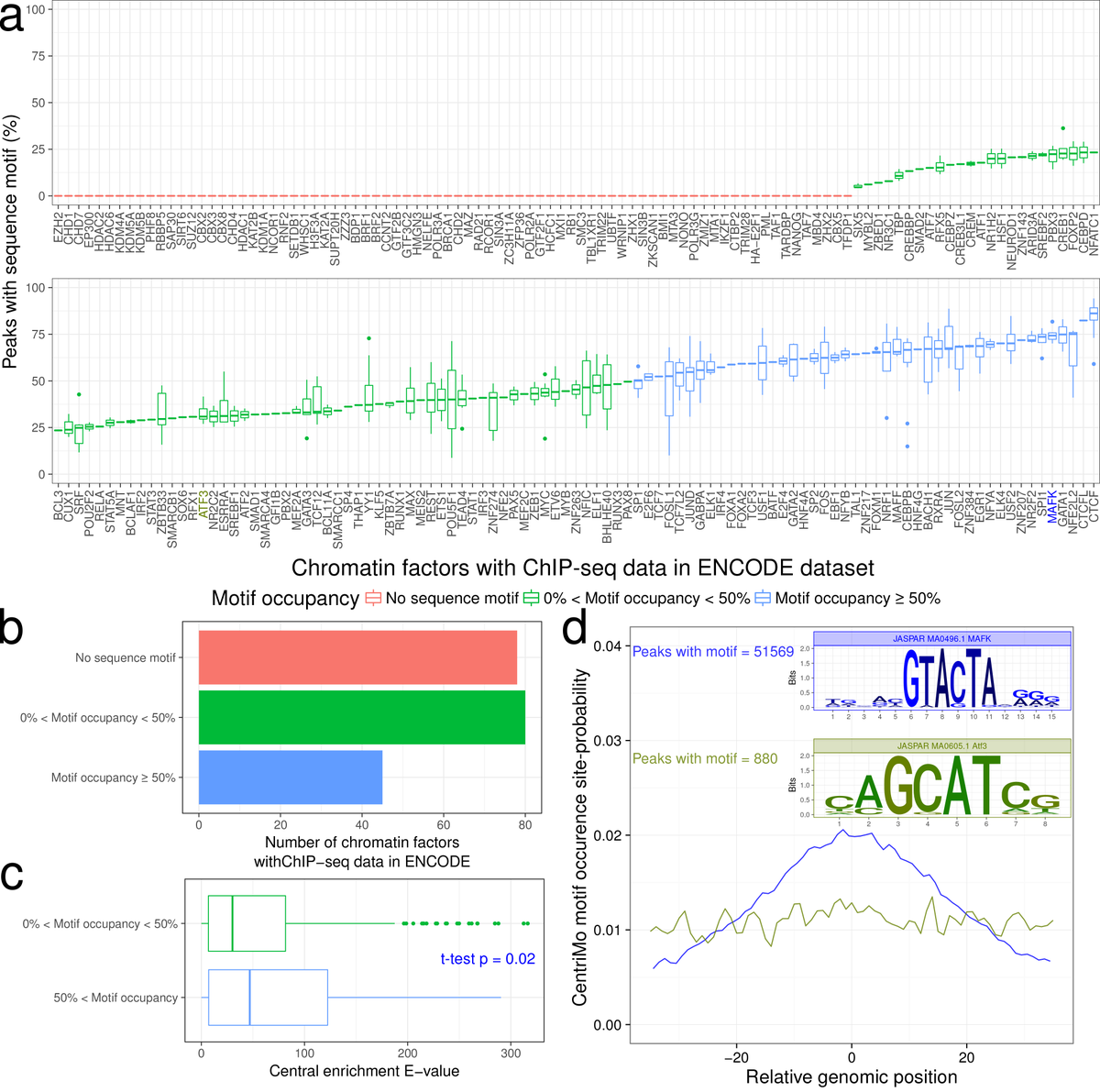
4/A crucial improvement in making better predictions came from making cell-type-specific predictions using data on open chromatin. These methods started with CENTIPEDE (@rogerpique @jkpritch) and the @AP_Boyle method in 2010. Summarized by Gusmao et al.: nature.com/articles/nmeth…
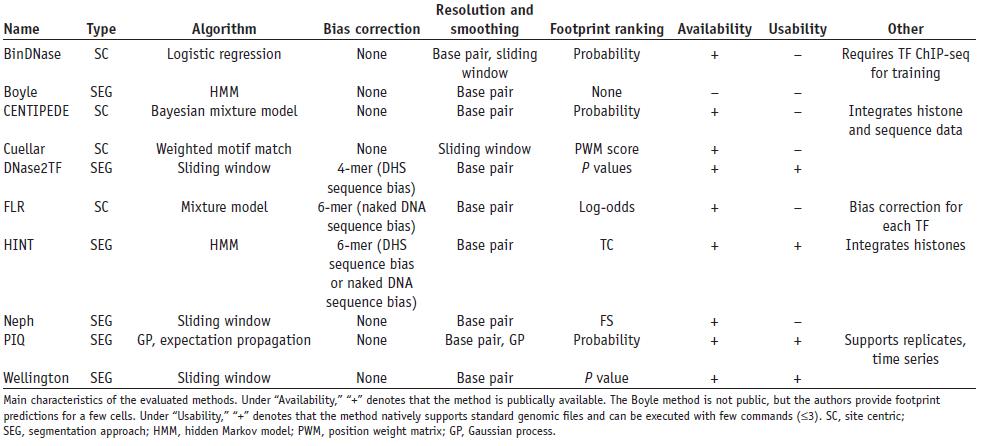
5/We wanted to improve predictions further and free them from a requirement of open chromatin footprinting found in some of these methods, as many chromatin factors will be present in places without open chromatin footprints.
6/There are many different ways to attack this problem, such as using ever-more-complex machine learning models. We decided to focus instead on reconceiving the problem.
7/What's the real-world scenario where one might use transcription factor binding prediction methods? It's definitely not the often-targeted case of having ChIP training data for some chromosomes and having to predict for others in the same cell type. Not going to happen.
8/A more likely scenario is that you want to predict transcription factor locations in a cell type for which you have a limited amount of sample. Enough to maybe run ATAC-seq and RNA-seq but not ChIP-seq for dozens of transcription factors.
9/In addition to this data for your new cell type, you have access to a whole universe of reference data in public databases, including ChIP-seq data on your transcription factor of interest in other cell types.
10/So what can we do with this data? First, we can build a model of how ChIP-seq signal in each 100-bp bin of the genome associates with global patterns of gene expression across multiple cell types.
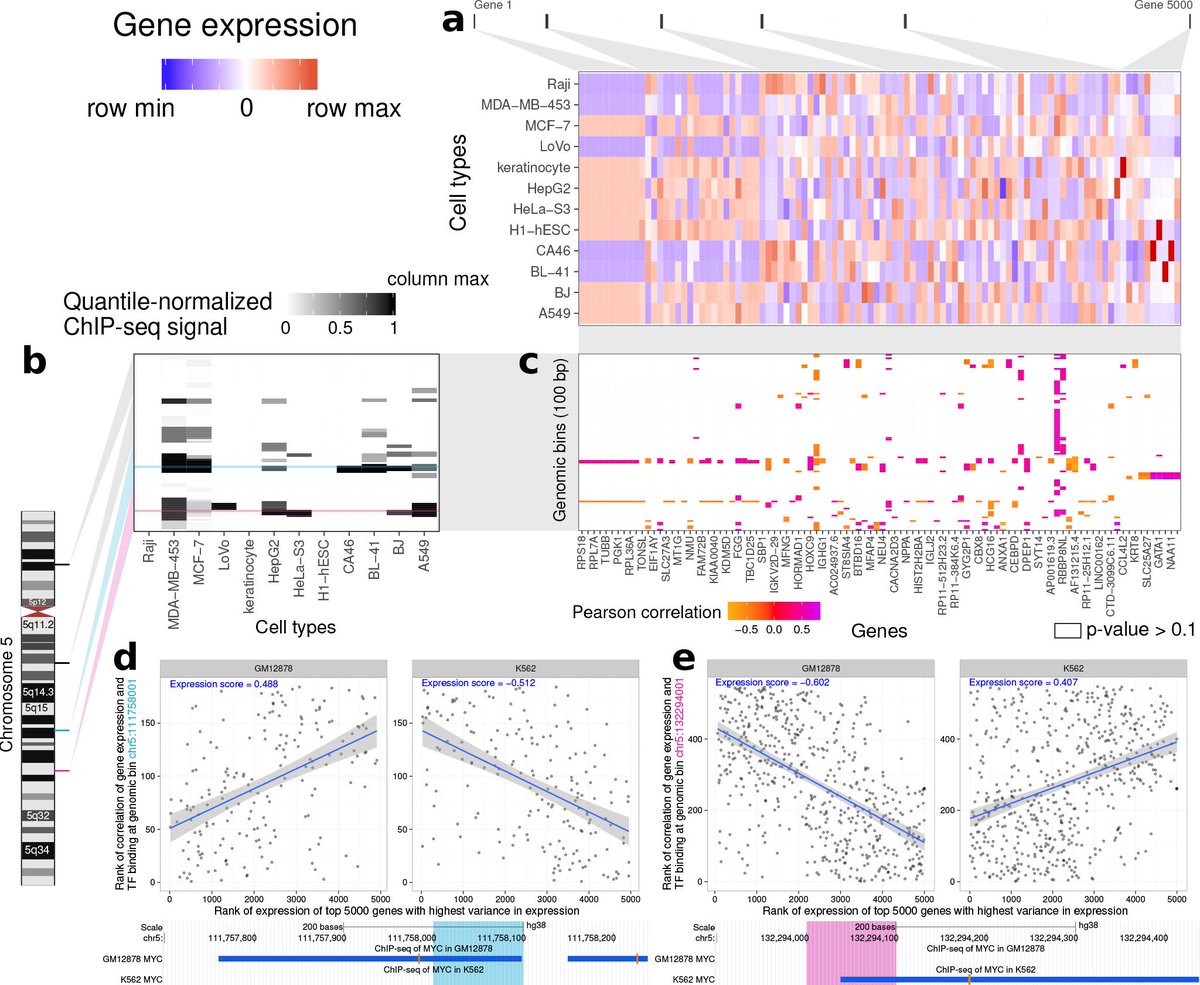
11/Many binding sites will associate with the expression of particular genes. Those genes can be anywhere in the genome and probably aren't near the binding site. Essentially, certain transcription factor binding patterns are associated with certain gene expression programs.
12/Second, we can use ChIP-seq data at a particular locus in a reference panel of cell types to influence our prediction of transcription factor presence at the same locus in the new cell type.
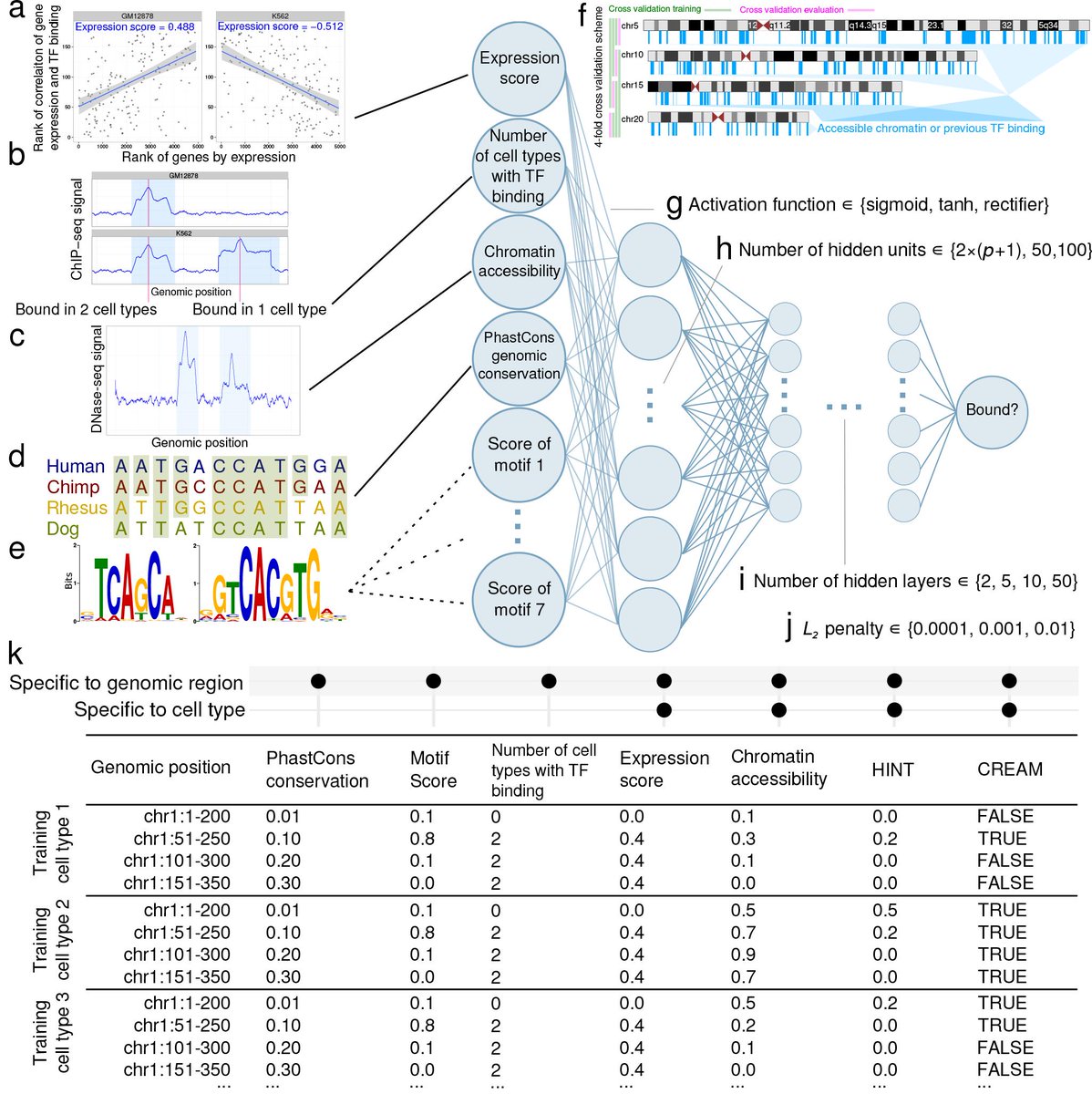
13/We combine these two new data types—the expression score and the number of cell types with TF presence at that locus—with other types previously used (chromatin accessibility, conservation, PWM score) as inputs to a neural network that predicts TF presence in a new cell type.
14/So how well does it work? For 10 chromatin factors it works extremely well, auPR > 0.5. This is exceptional performance for this task. Another 28 chromatin factors have auPR > 0.25.
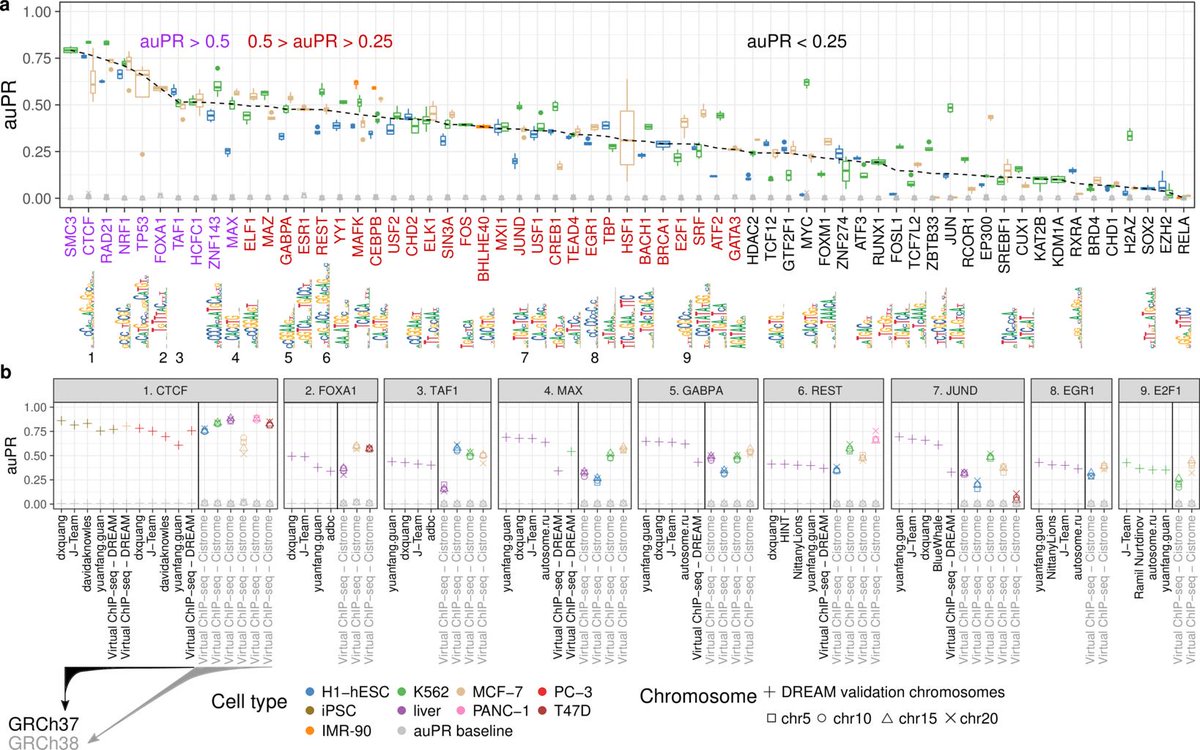
15/The remaining 25 chromatin factors have auPR < 0.25 which leaves much room for improvement. Still probably much better than other widely-used approaches such as only looking at PWM scores. In fact, 11 of these 25 don't have motifs so the PWM approach isn't even possible.
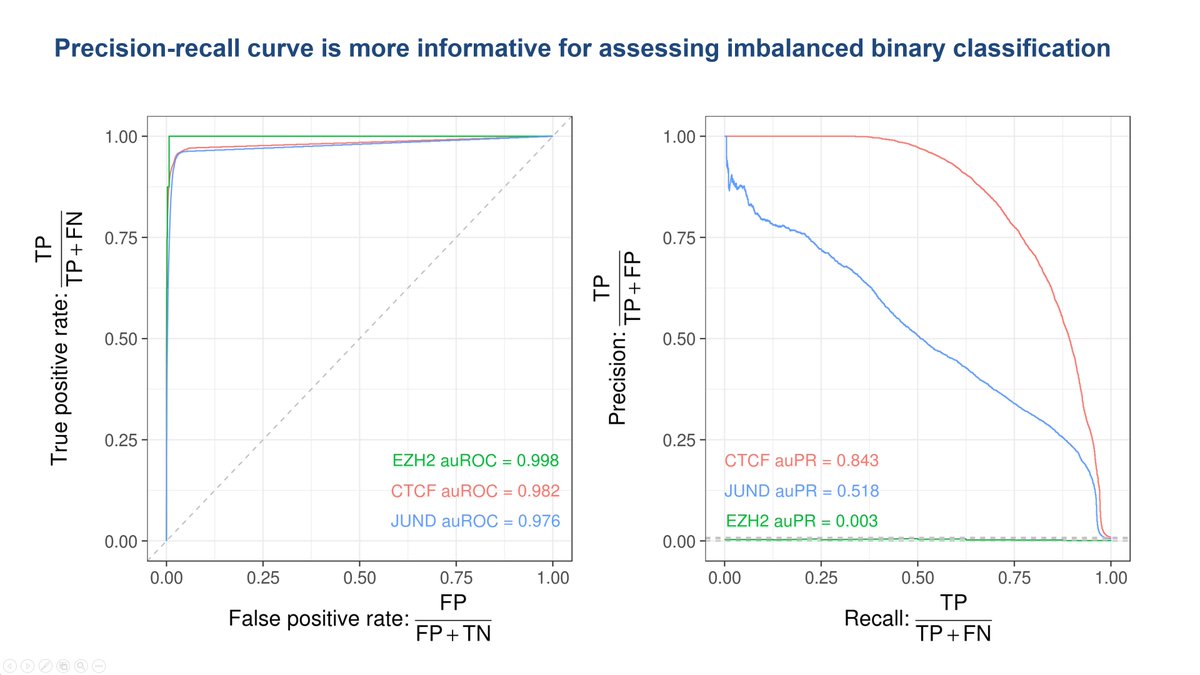
16/We measure performance here with auPR, not the auROC widely used in the past. Our second-worst model for EZH2 actually has auROC of 0.998! But I don't advise relying on it. For most genomic classification tasks, it's time to leave this inflated and misleading metric behind.
17/For our best 36 chromatin factors, we report a variety of benchmarks, including auPR, auROC, F1, accuracy, and Matthews correlation coefficient. You should always require multiple performance benchmarks. The trade-offs for each are different and can be instructive.
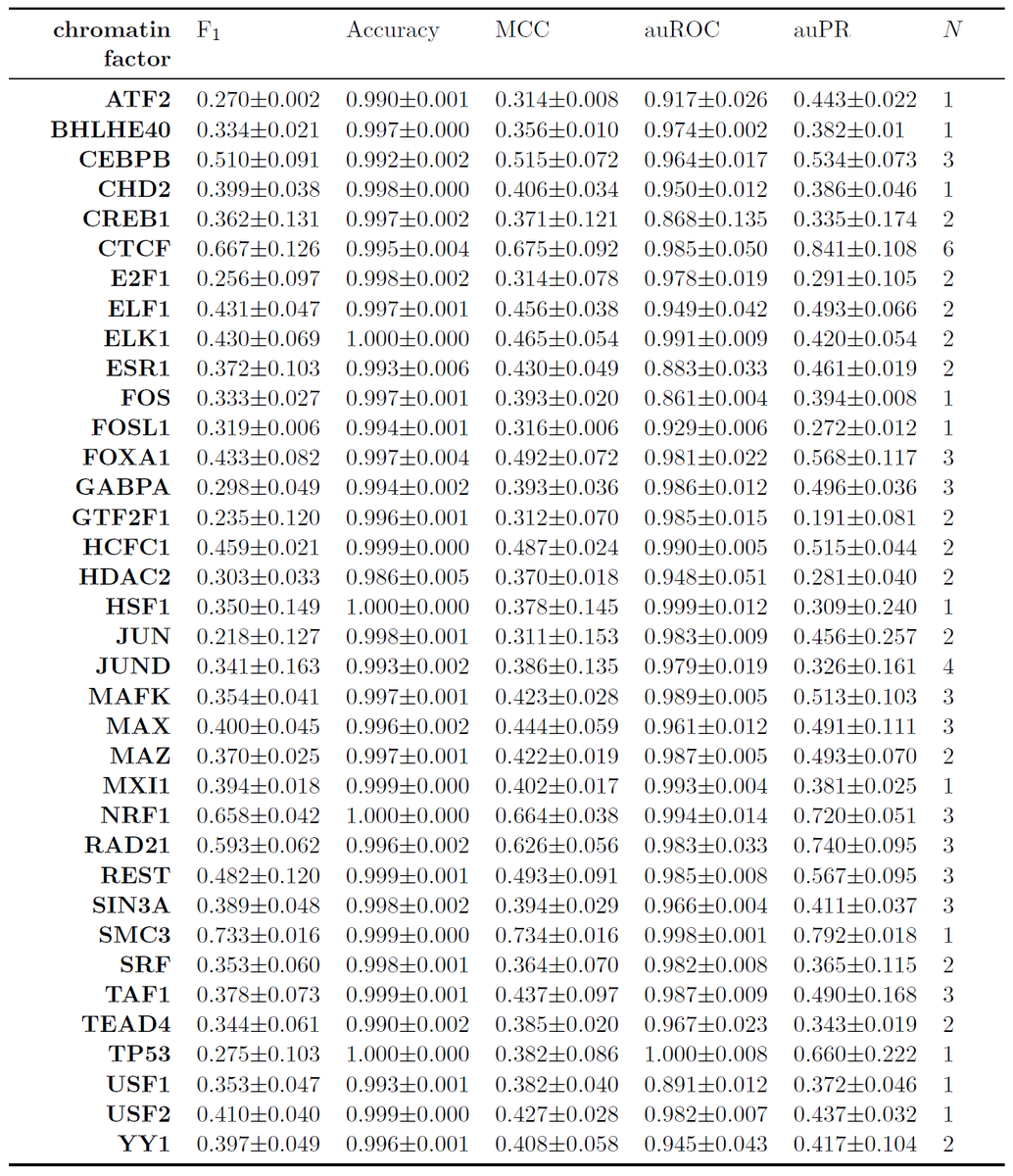
18/We compared against the best performers in the @ENCODE_NIH-@DR_E_A_M in vivo TF binding site prediction challenge. Unfortunately, it's not really possible to do an apples-to-apples comparison that's fair for everyone. synapse.org/ENCODE
19/First, the whole thesis of our approach is that in most real-world scenarios for TF prediction you'll be able to use things like reference ChIP-seq data in other cell types. DREAM Challenge participants were expressly forbidden from using these data. synapse.org/#!Synapse:syn6…
20/Second, the limited ChIP-seq data in the DREAM Challenge meant we often didn't have enough to use our ability to its full potential. So we couldn't compare against them comprehensively.
21/You can see the comparisons we made in the figure above. Keep in mind auPR is highly dependent on the test set used, so one cannot directly compare across different test sets, emphasized by the vertical lines within panel b.
22/We looked further into what data features associate with correct or incorrect predictions. Some of these models produce more false negative predictions than false positive (left). The others produce more false positives.
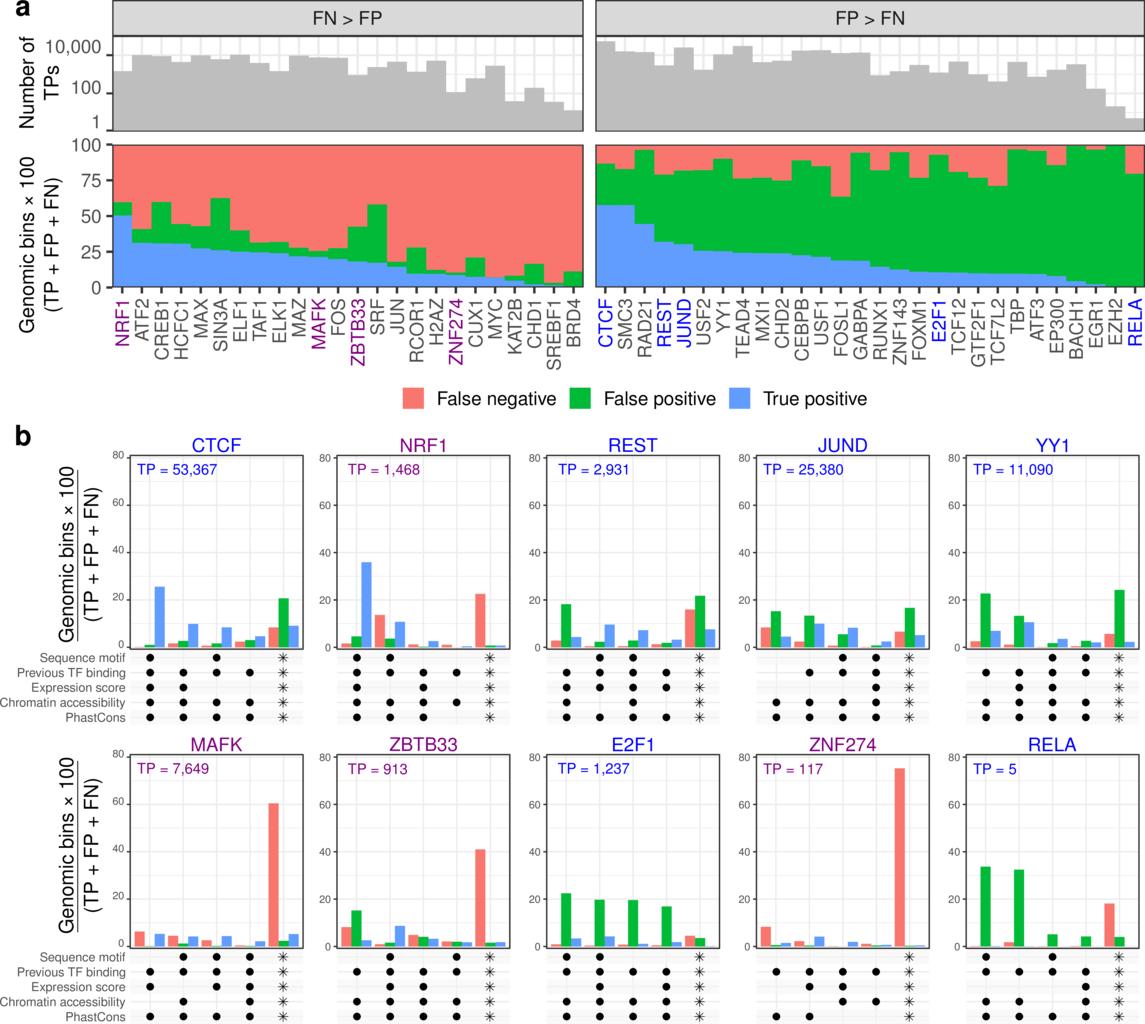
23/Looking into what features are most important for correct predictions, we find that varies a lot by model. For many, chromatin accessibility is essential, but for others it's not (like ATF2, REST, MAFK).
24/Presence of some chromatin factor in another cell type is found for most correct positive predictions. Still, for some chromatin factors, such as TEAD4 and GABPA, we made correct positive predictions without this signal.
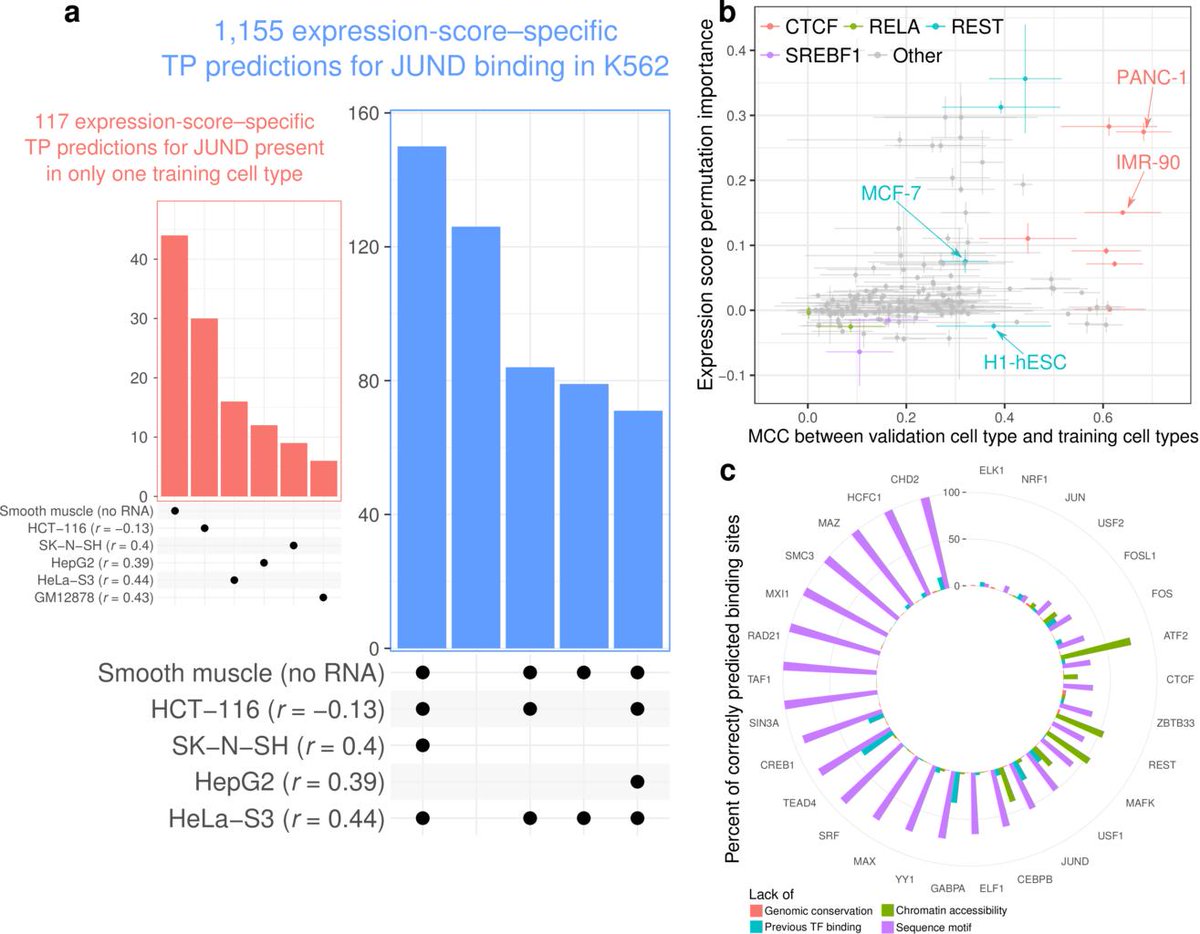
25/To see which input features were most important, we tried training our models with only subsets. The reference ChIP-seq data in other cell types and the expression score were the most important features. Chromatin accessibility and sequence motifs usually helped but not always
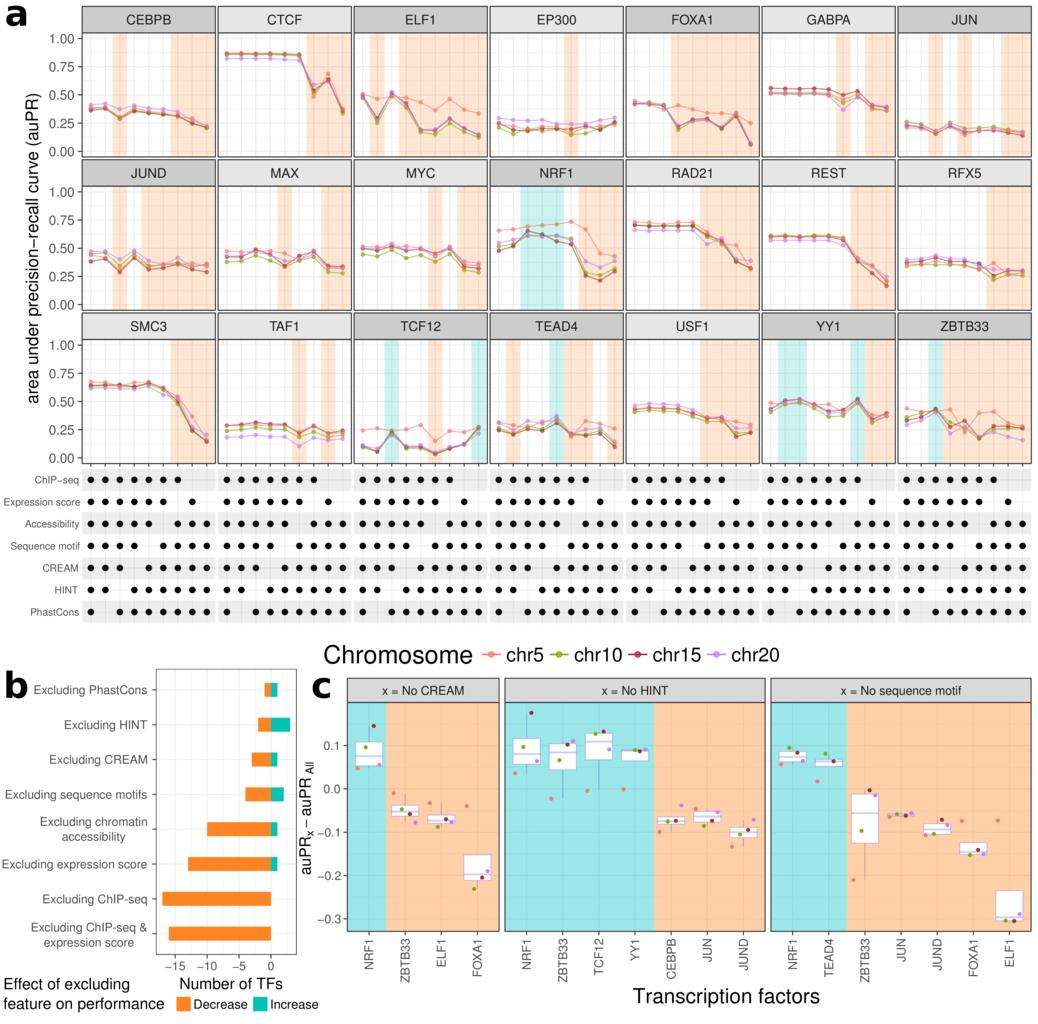
26/Incorporating output of integrative methods like HINT or CREAM resulted in what appeared to be overfitting. These methods are definitely useful but since our model already have the underlying data these models use (such as open chromatin data) they don't seem necessary here.
27/We also looked further into why the expression score works. Our hypothesis is that genes whose expression follow the same trends the binding of some chromatin factor are involved in the same regulatory processes.
28/We accurately cluster the chromatin factors into functional categories based on the genes whose expression the factor's binding relates to.
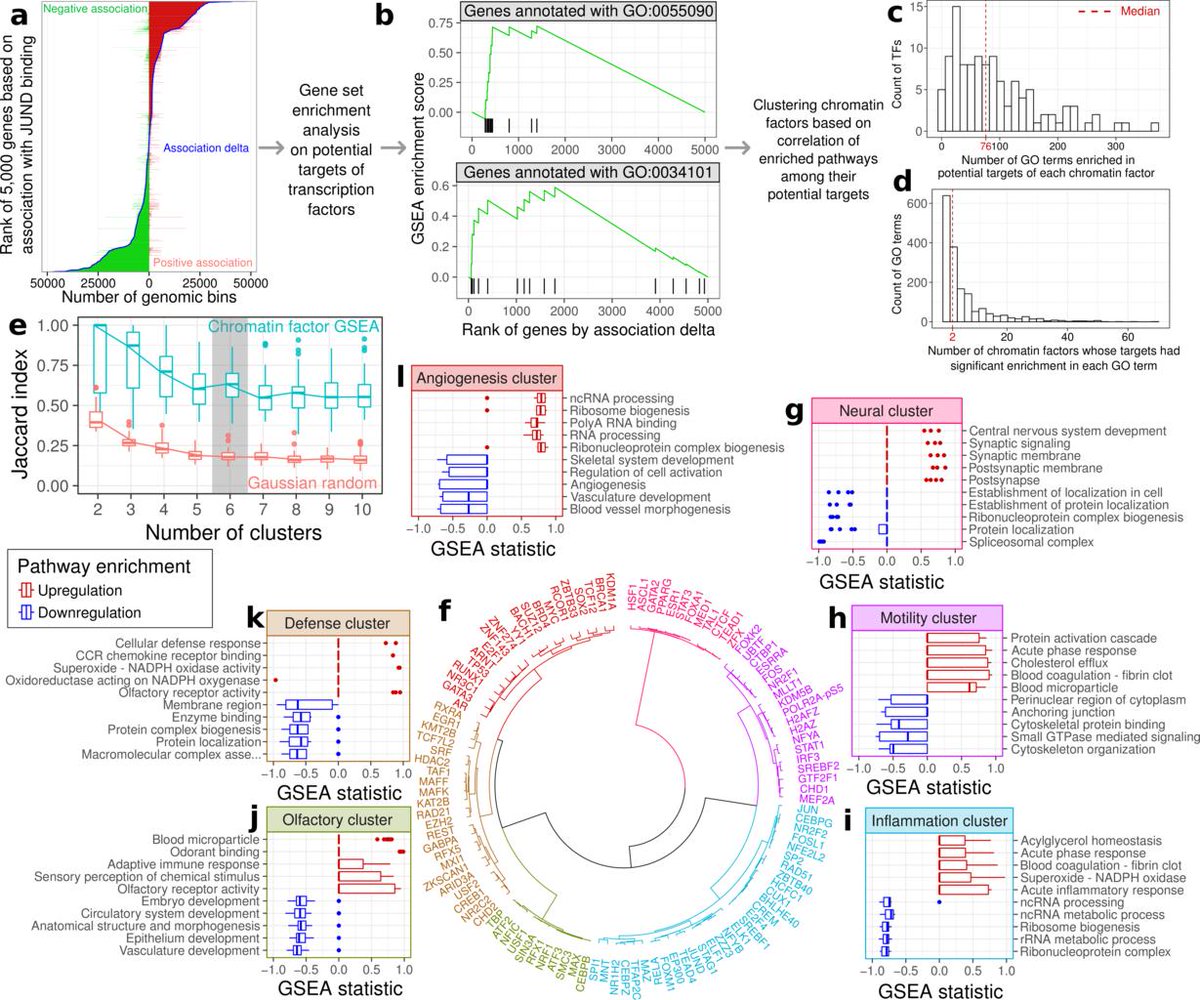
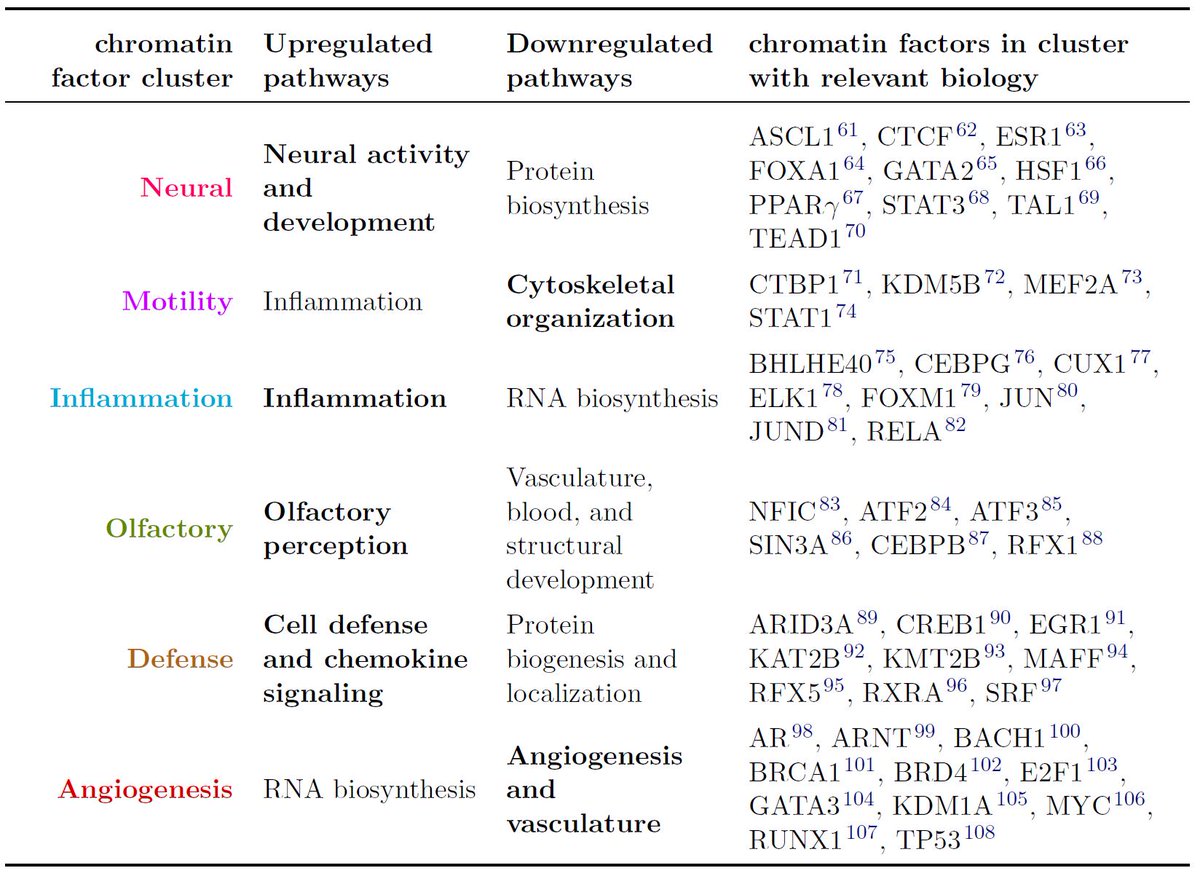
29/Finally, we've used our best-performing 36 chromatin factor models to make predictions on 33 different Roadmap Epigenomics human tissue types. You can load these in @GenomeBrowser just by visiting virchip.hoffmanlab.org and clicking the "Virtual ChIP-seq track hub" button.
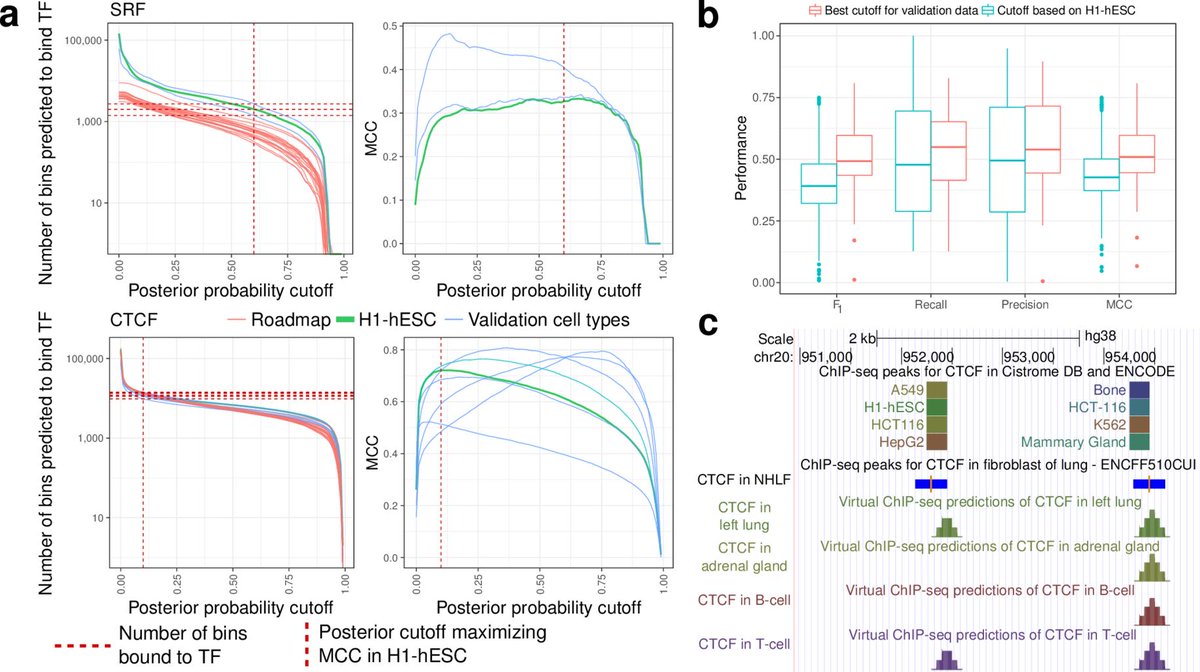
30/Within @GenomeBrowser, you can then turn enable the display of predictions for any of these 36 factors.
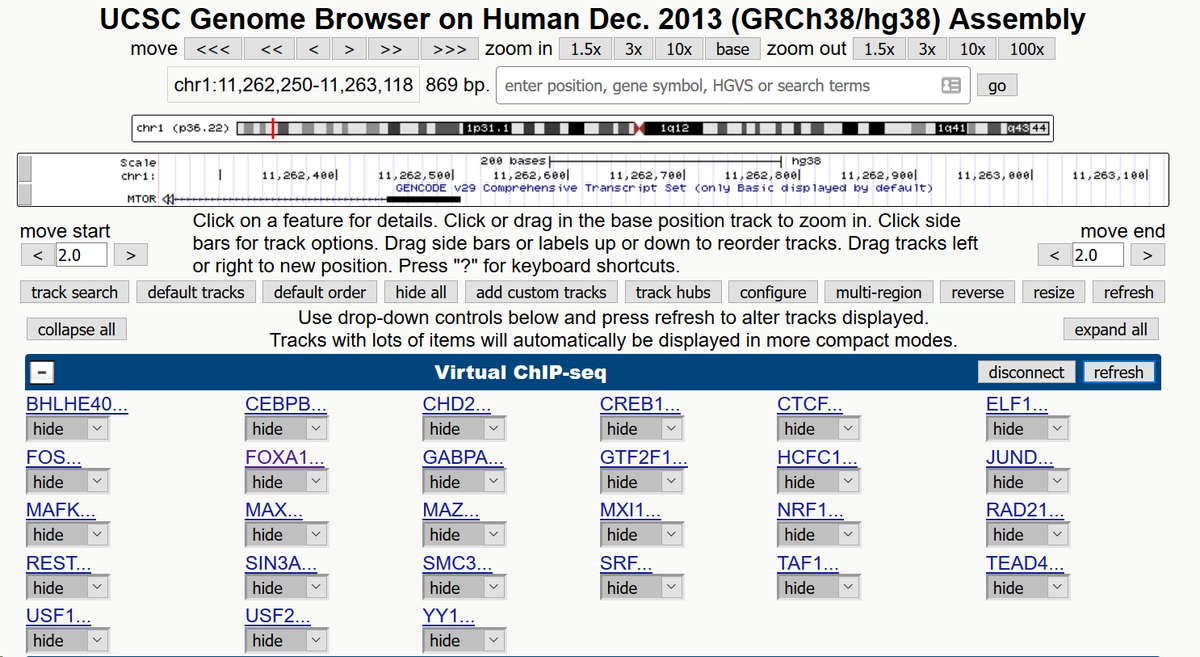
31/You can then turn on tracks for whatever tissue types you're interested in.
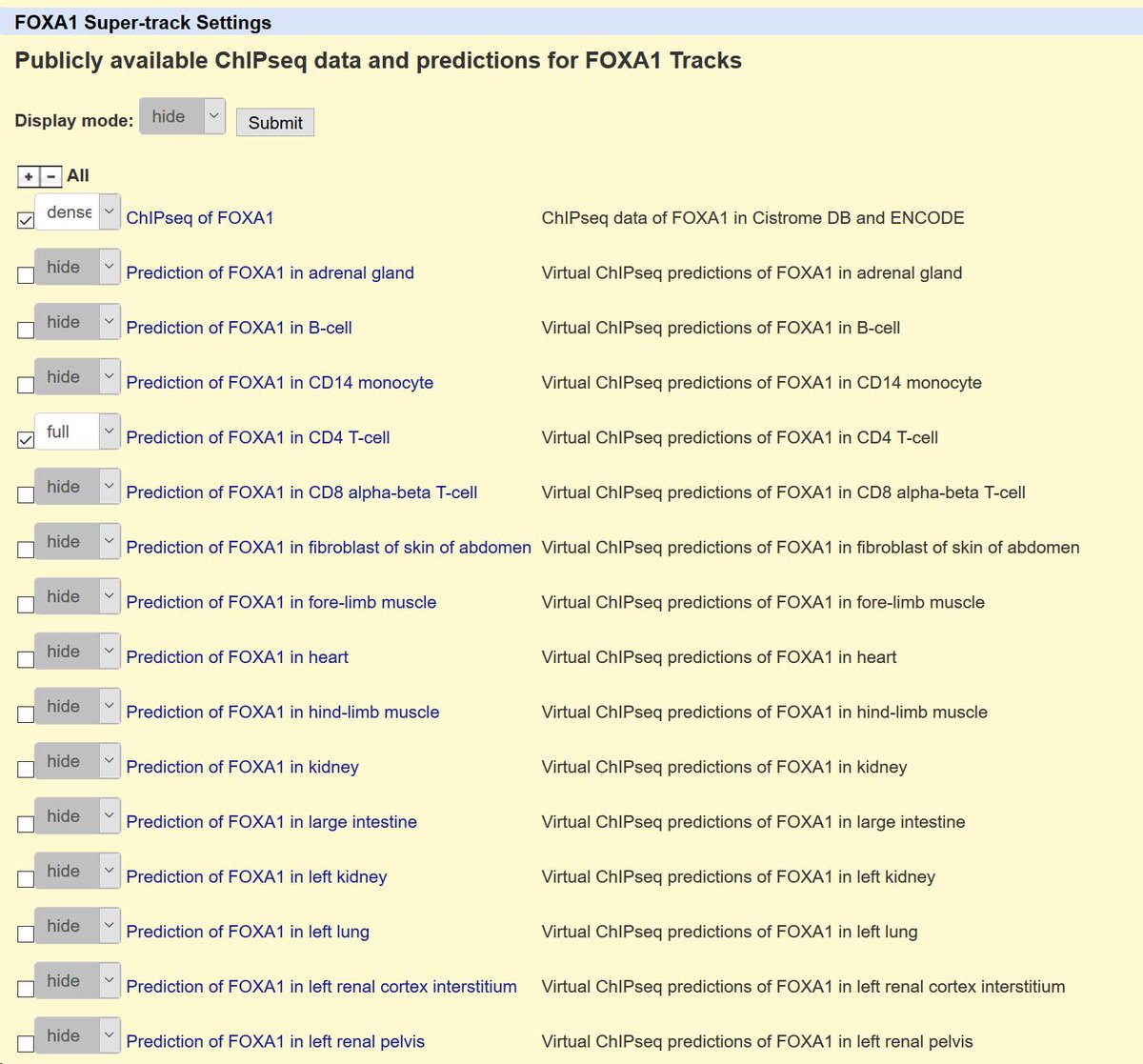
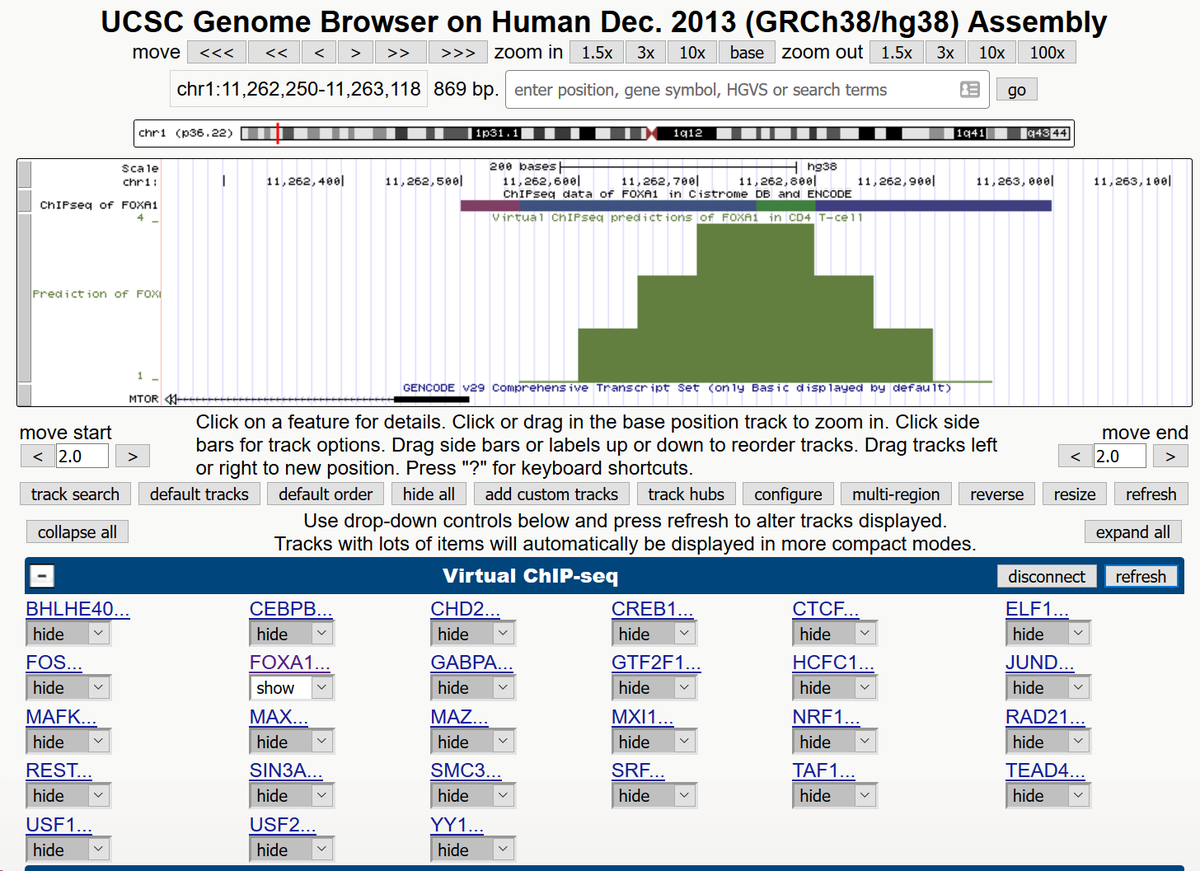
32/Voila, prediction of a FOXA1 binding site in the MTOR promoter in CD4 T-cells. No programming necessary. You don't even have to run the software yourself—all these predictions are ready for you to use now.
33/Of course, if you do want to run the software, it's available on @Bitbucket. Let us know how you find it! The software is also available on @ZENODO_ORG, as are our predictions, and the datasets we used to train and test the models. bitbucket.org/hoffmanlab/vir…