Solid paper by @MKarimzade and @michaelhoffman . Some comments to follow.
In summary, the paper proposes a powerful imputation based approach for predicting TF binding in NEW cell types. Note that this is the practical prediction problem. Not cross-validation, where u simply hold out chromosomes in cell types where u already have TF ChIP-seq data 1/
Prediction in new cell types is the scenario we proposed to evaluate in the ENCODE-DREAM TF binding challenge. Kudos to the authors for evaluating using this formulation which is the practical scenario for using the predictor. 2/
Worth noting that almost all other papers in the literature that claim to 'predict' TF binding, do not evaluate predictions in new cell types. I know I am repeating myself. But this is a CRITICAL piece of the problem design that almost everyone seems to ignore. 3/
Next, the authors evaluate performance chromosome wide using a performance measure that is sensitive to the class imbalance. This is also how we evaluate performance in the ENCODE-DREAM challenge. This is again a CRITICALLY important piece of evaluation of performance. 4/
I harp about this incessantly. Your predictor will be used in practice to scan entire chromosomes. Hence, you MUST evaluate your predictor on ENTIRE chromosomes. 5/
Kudos to the authors for evaluating performance 'CHROMOSOME WIDE IN HELD OUT CELL TYPES (sorry for type-shouting in all caps. Can't stress how important this is) 6/
Now, lets discuss the evaluation measures the authors used. As we did in the ENCODE-DREAM Challenge, they stress the importance of using performance measures that are sensitive to class imbalance. 7/
This is also CRITICAL because across an entire chromosome/genome, a very small fraction of nucleotides will ever be bound by a TF. That means there is very significant imbalance between the number of bound sites and the number of unbound sites. 8/
The auROC which is the most commonly used performance measure in most publications, is insensitive to class imbalance. Basically it tells u the probability that a random bound site will be ranked above a random unbound site. 9/
In practice we don't care about this probability. The auROC compares the true positive rate (TPR) to the false positive rate (FPR) of the predictor at different thresholds on the predictor's scores. But we dont really care about the FPR for these unbalanced problems. 10/
We care about trading off the true positive rate (TPR) against the FALSE DISCOVERY RATE (FDR) which is (1-precision). Note that FDR is not the same as FPR. See definitions here en.wikipedia.org/wiki/Receiver_… 11/
The authors hence use several measures that are explicitly sensitive to TPR and FDR such as the area under the precision recall curve, F1 scores and MCC. 12/
This is important because for problems with significant class imbalance, you can get fooled by high auROCs that translate to terrible TPRs at desired reasonable FDRs (e.g. 10-50%) 13/
The authors, do however, tout the raw accuracy of the classifier in the abstract "This approach .... predicts binding for 34 transcription factors (accuracy > 0.99; Matthews correlation coefficient > 0.3)". This is unnecessary IMHO. @michaelhoffman @MKarimzade 14/
An accuracy of >0.99 sounds incredible. But like the auROC, for the imbalance in the data, it is just not very useful. A classifier that trivially always predicts unbound will have an accuracy close to 0.95 - 0.99 for TFs. 15/
Might as well highlight the measures we actually care about such as the MCC, PR at X% FDR, auPR etc. 16/
Now, onto the method itself. Several novel contributions here. The authors propose a novel expression score associating binding activity of the target TF in any region of the genome with expression levels of any gene across a collection of reference training cell types 17/
The authors then train a neural network to predict binding at any region in a held out cell type using multiple input features of the region including the expression score of the region in held out cell type, TF motif features, chromatin accessibility, conservation. 18/
The auPR of their models for various TFs range from ~0.1 to ~0.8 with a majority in the range between 0.25 and 0.5. These are very significant improvements over the state of the art. 19/
It still worth noting that these numbers are very different from the perception of performance u get when u look at raw accuracy (> 0.99) or auROCs (>0.9 in most cases). 20/
Its also worth noting that the auPR of a random classifier on a dataset with a class imbalance of 1% is 0.01. Most TFs exhibit a class imbalance lower than this. So the predictor the authors have developed (auPR 0.1-0.8) is doing very well. 21/
However, also worth noting that these auPRs with translate to probable TPRs in the range of ~20-30% at 50% FDR. So this is far from a solved problem. But this is the reality of how hard the problem is. 22/
Another important point the authors make is that the sequence affinity (motifs) of the TF which is what most previous methods have focused on is far from being the most predictive feature. We will elaborating a lot more on this in our DREAM Challenge paper. 23/
This observation does not mean that sequence does not matter for binding. It absolutely does. But relying on the canonical motif of the target TF only is insufficient. 24/
Infact, if you modeled all other sequence features (e.g. co-factor motifs, GC content, whatever u like) that are predictive in your training cell types (which the authors do not), this will only hurt you more when predicting in a new cell type. Thought exercise. Why? 25/
Also worth noting that in this 'imputation' formulation of the problem used by the authors, sequence features do not appear very important CONDITIONED on using the 'expression score' 26/
The expression score of a region as we noted implicitly encodes the observation of a TF binding event at that region in a reference training cell type AND the similarity of that reference training cell type to the test cell type (using expression). 27/
You can achieve comparable/better performance than this formulation using just sequence+DNase, without using the expression score. But you need to model sequence very cleverly. And in this alternate formulation, sequence is important. More on this in the DREAM challenge paper 28/
This is where I will raise a few issues with some of the comparisons reported in the paper relative to the DREAM Challenge winners 29/
A statement in the paper says "For CTCF, FOXA1, TAF1, and REST, Virtual ChIP-seq had a higher auPR in at least one validation cell type than any DREAM Challenge participant" 30/
Another statement says "for CTCF and REST, Virtual ChIP-seq outperformed any other challenge participant in the same cell type evaluated in the final round of the challenge (Figure 4b)" 31/
Lets examine these statements in more detail 32/
Lets look at the CTCF panel in figure 4B. The first 4 columns are the DREAM challenge winners. The last 2 columns are virtualChIPseq. The color represents the test cell type. Keep an eye on the colors/cell types. They are important in this discussion 33/ 
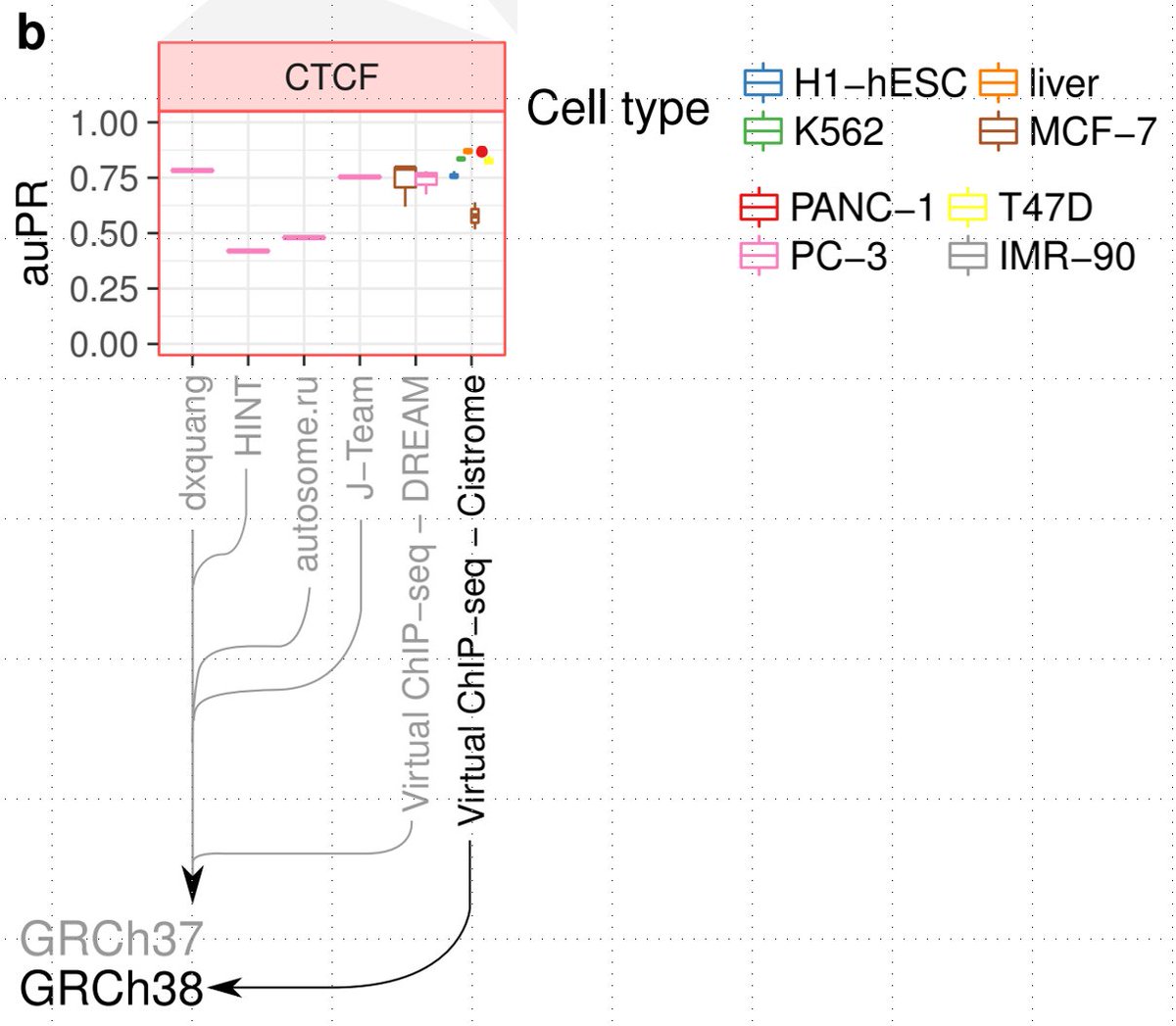
The highest performing model amongst the DREAM winners for CTCF in PC-3 (light pink cell type) is dxquang (first column). You can see that on the same cell type (light pink), second last col. Virtual ChIP-seq trained on DREAM data in fact does worse than dxquang 34/
VirtualChIPseq in last column (trained on cistrome data) seems to have higher auPR than dxquang for several cell types (several colors). BUT, these are not the same cell type as the ones in the first 4 columns (the last column does not have a light pink point). 35/
You cannot compare auPR of two methods for the same TF in different cell types. Class imbalance, quality of data/labels differs across cell types etc. making auPRs not directly comparable. 36/
You can only compare auPR of two competing methods for the same TF in matching test cell types on matching test chromosomes w/ identical ground truth labels. 37/
I'm not sure why the authors do not report a pink point (ie predict in PC-3) in the last column. As I noted, dxquang (col1) does beat VirtualChIPseq (col 4) on matched test cell types (pink PC-3). 38/
Also test chromosomes are (likely) not the same across the two methods. In DREAM, we used chr1, 21, 8 as test chr (with chr8 as the true blind test chr). VirtualChIPseq uses a different set of chrs for evaluation. 39/
The same is true for other TFs in Fig 4B as well. I see no TF for which VirtualChIPseq beats the top performing DREAM winner on matching test cell types. In fact, for matches test cell types, VirtualChIPseq does slightly worse or substantially worse than the top DREAM winner 40/
VirtualChIPseq is doing very well. I'm simply pointing out that the specific claims about beating the DREAM winners are not valid (as I perceived them) from Fig 4B. @MKarimzade @michaelhoffman let me know if I have misunderstood something here. 41/
One nitpicky thing, "For most of these TFs, the DREAM Challenge held out test chromosomes instead of test cell types." This is incorrect. 42/
In the DREAM Challenge, for ALL TFs, participants were expected to predict in held-out chromosomes in held-out cell types. 43/
This is in fact a harder problem. We were testing generalization of the predictors to new cell types and to new sequences (chromosomes) they had never seen before in training. We also evaluated performance genome-wide in the new cell types 44/
Moving on, the authors then use VirtualChIPseq to predict binding in several important primary cell types and tissues from the Roadmap Epigenomics project (samples in which it would be very difficult to impossible to perform high quality TF ChIP-seq). 45/
Overall, an excellent paper with strong performance, sound evaluation metrics and a great resource. 46/
The only part I am not convinced about are the claims that the method is outperforming the DREAM winners on any of the TFs on matched cell types. But this is a relatively minor concern. 47/
I also want to make a prediction. With clever modeling of sequence and better ensembling (will elaborate more on this soon), obtain substantially better models than what we see here or in the DREAM challenge. 48/
But I hope this paper and the imminent DREAM challenge preprint will serve as benchmark datasets, benchmark models and practical, realistic evaluation guidelines for further studies claiming to 'predict' TF binding. 49/49
Oh and one more thing. ChIP-seq has tons of artifacts. Its the best we got at the moment to measure invivo binding. I estimate that we are underestimating our 'true performance' because of the issues with the 'ground truth' based on ChIP-seq. 50/
But also that the 'true performance' of these and any other current models is in the literature are nowhere even close to something one would consider reasonable (e.g I would consider >60% recall at 10% FDR as a high performance model). 51/
Thanks for reading this ultra-long thread. 52/52