Proposed COVID-19 Pathophysiology [Part 3 of (now) 4]:
Coagulopathy Strikes Back.
What happens when #COVID19 clashes with the world of hematology?
Buckle up again for a long one.
...let’s dive in like this macrophage (exploring all directions):
Coagulopathy Strikes Back.
What happens when #COVID19 clashes with the world of hematology?
Buckle up again for a long one.
...let’s dive in like this macrophage (exploring all directions):
First, what do you think is the predominant mechanism by which thrombosis occurs in COVID-19?
Where does thrombosis typically first occur?
Why does thrombosis occur?
As @HermelinMD beautifully points out in her many tweetorials, pathology is the lens through which we can glean an immense amount about the underlying causes of disease (microscopy pun intended).
...so what have we seen pathologically in COVID19?
...so what have we seen pathologically in COVID19?
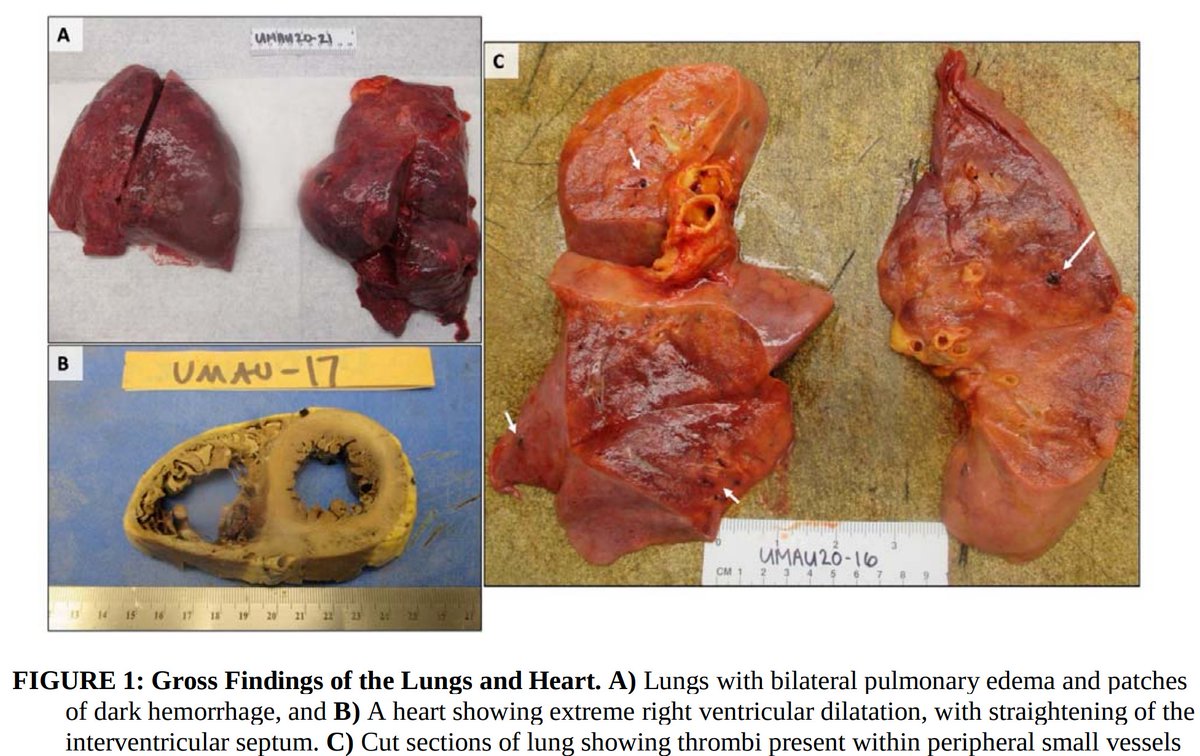
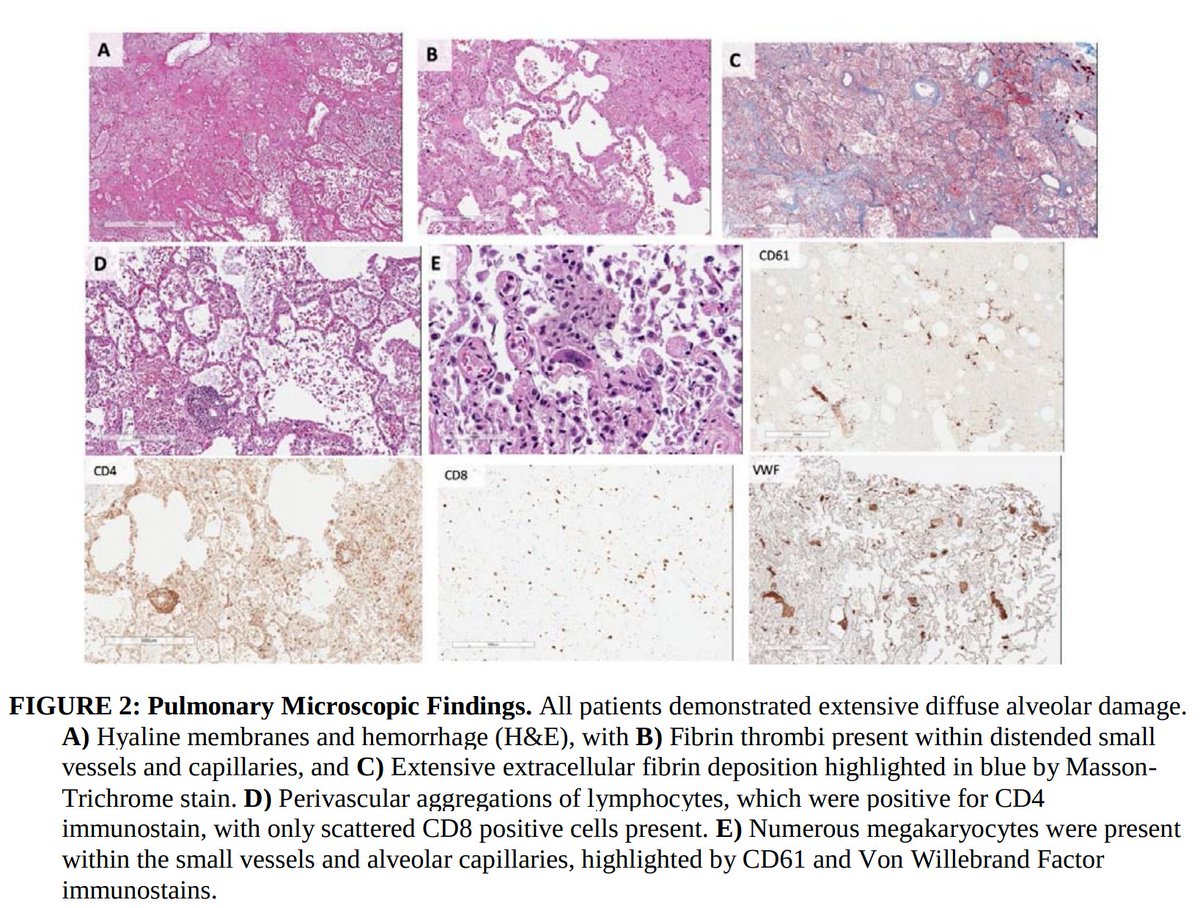
1. Pulmonary microvascular thrombosis with diffuse alveolar damage (DAD).
Early reports described “marked inflammatory changes including mononuclear cell infiltrates, virally infected cells, and DAD”.
2. Increased VTE incidence, with both microvascular thrombosis and PE.
Early reports described “marked inflammatory changes including mononuclear cell infiltrates, virally infected cells, and DAD”.
2. Increased VTE incidence, with both microvascular thrombosis and PE.
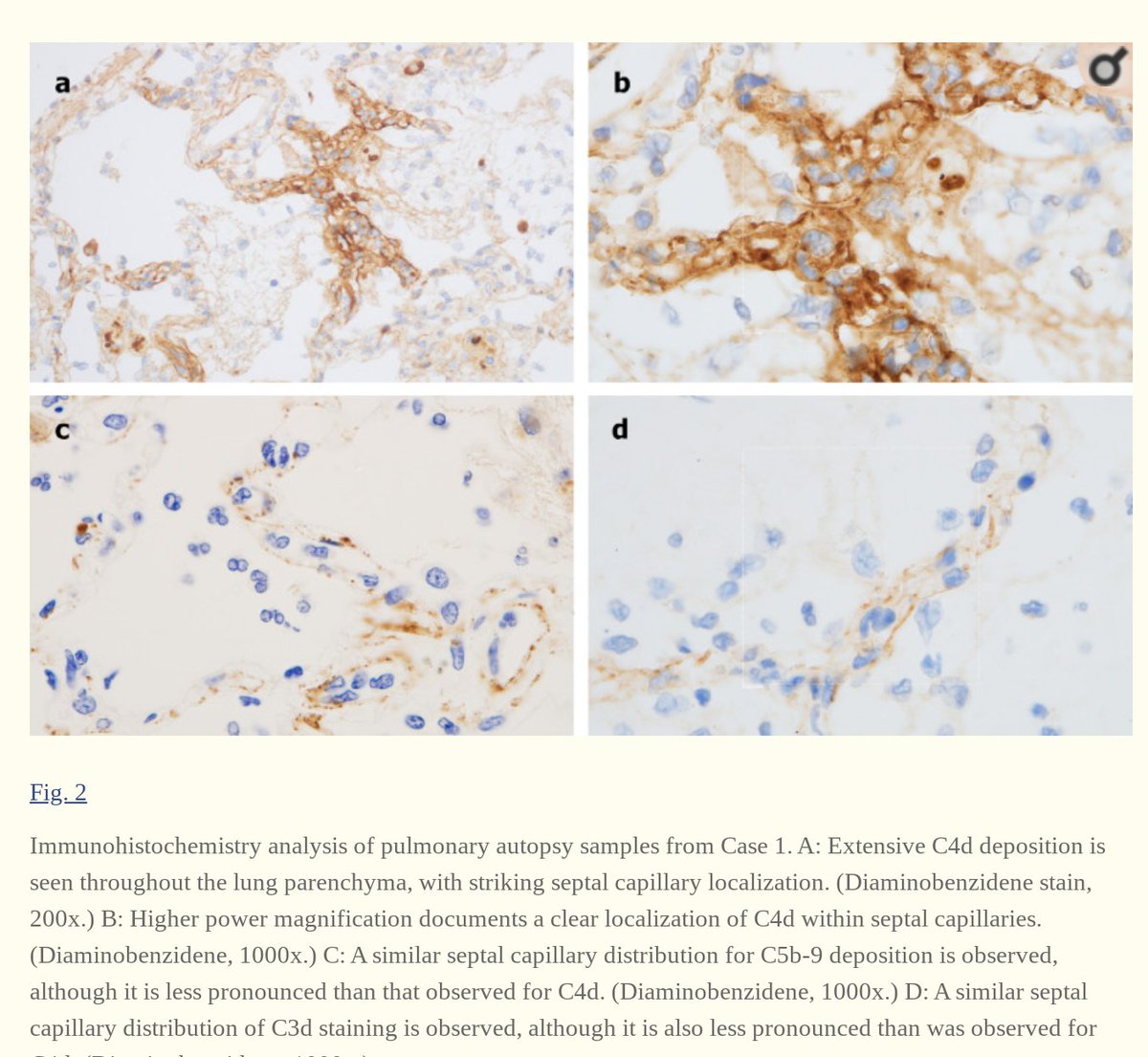
3. Complement deposition, with a striking localization to the capillary beds.
Notably, co-localization of SARS-CoV-2 envelope proteins has been seen with complement components in dermal tissues and pulmonary microvessels.
Notably, co-localization of SARS-CoV-2 envelope proteins has been seen with complement components in dermal tissues and pulmonary microvessels.

So do these findings reflect overt DIC? Or is something else at play?
First let’s review what overt DIC looks like:
First let’s review what overt DIC looks like:
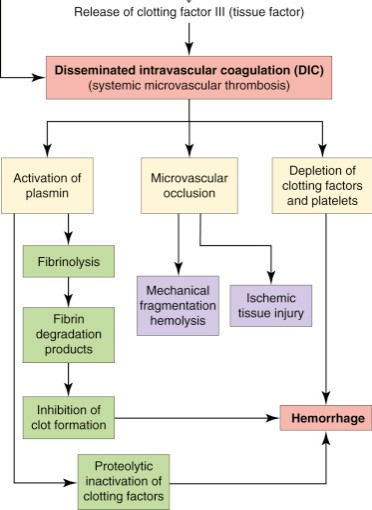
In identifying a disease phenotype, what is not seen can often be just as important as what is observed.
What in the above diagram aren’t we seeing in COVID and how does it contrast to DIC?
What in the above diagram aren’t we seeing in COVID and how does it contrast to DIC?
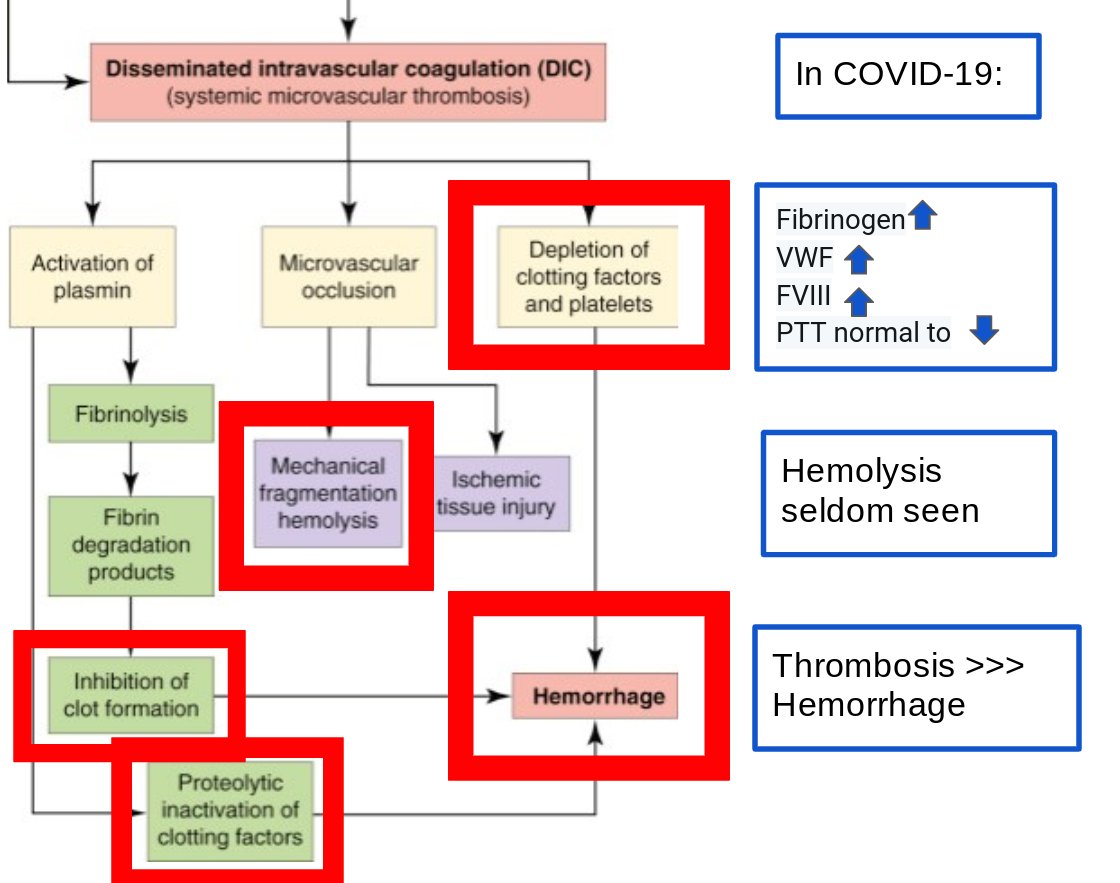
Since the outset, thrombosis in COVID is frequent:
Cui et al (Chinese): 81 ICU pts, 25% VTE incidence without VTE ppx
Kiok et al (Dutch): 184 ICU pts, VTE in 27% despite ppx AC
Llitjos et al (French): 26 ICU pts, VTE in 69% of pts on therapeutic (56%) or prophylactic (100%) AC
Cui et al (Chinese): 81 ICU pts, 25% VTE incidence without VTE ppx
Kiok et al (Dutch): 184 ICU pts, VTE in 27% despite ppx AC
Llitjos et al (French): 26 ICU pts, VTE in 69% of pts on therapeutic (56%) or prophylactic (100%) AC
(For context, the usual failure rate of AC ppx in ICU pts is ~7.7%)
With such an increased propensity for thrombosis, increased D-dimer levels have also been correlated with various risks in COVID-19:
With such an increased propensity for thrombosis, increased D-dimer levels have also been correlated with various risks in COVID-19:
1. D-dimer > 3.0 mg/L + routine heparin ppx = ⬇️ mortality (Tang et al)
2. Progressive⬆️over 7 days following admission is seen in non-survivors
3. >1 mg/L = HR of 18.4 for in-hospital mortality
4. 3-4x ULN D-dimer at baseline = need to admit to hospital (per ISTH guidelines)
2. Progressive⬆️over 7 days following admission is seen in non-survivors
3. >1 mg/L = HR of 18.4 for in-hospital mortality
4. 3-4x ULN D-dimer at baseline = need to admit to hospital (per ISTH guidelines)
But what does an elevated D-dimer level fundamentally tell you?
Fundamentally, ⬆️D-dimer reflects the presence of fibrinolysis.
uPA and tPA release from endothelial cells cleaves fibrin into split products.
As fibrin is already cross-linked (thanks to FXIII), they are cleaved as dimers.
And the largest, readily measurable dimer? D-dimer.
uPA and tPA release from endothelial cells cleaves fibrin into split products.
As fibrin is already cross-linked (thanks to FXIII), they are cleaved as dimers.
And the largest, readily measurable dimer? D-dimer.

So is this reflective of overt DIC?
...probably not.
Marongiu et al suggest rather than overt DIC, a localized intravascular coagulation (LIC) first occurs in the lungs.
This moreso reflects thromboinflammation, or the complex interplay between acute inflammation & thrombosis.
...probably not.
Marongiu et al suggest rather than overt DIC, a localized intravascular coagulation (LIC) first occurs in the lungs.
This moreso reflects thromboinflammation, or the complex interplay between acute inflammation & thrombosis.
Have you heard of thromboinflammation?
If no, then good, because now you’re learning something.
First, true or false:
Thrombosis can be a protective mechanism in the setting of infection.
First, true or false:
Thrombosis can be a protective mechanism in the setting of infection.
It’s true!
Though thrombosis is seldom discussed as an immunological phenomenon, the formation of fibrin clot can trap circulating bacteria and help in their phagocytosis, antigen presentation, and ultimately, eradication.
Think of a fish net catching salmon on a river.
Though thrombosis is seldom discussed as an immunological phenomenon, the formation of fibrin clot can trap circulating bacteria and help in their phagocytosis, antigen presentation, and ultimately, eradication.
Think of a fish net catching salmon on a river.
Speaking of nets, neutrophils (PMNs) can act as fishermen/fisherwomen (better than guys above)
PMN NETs (neutrophil extracellular traps) occur as the neutrophils rapidly extrude their intracellular DNA, leading to a proteinaceous net that helps trap & kill bacteria (see below)
PMN NETs (neutrophil extracellular traps) occur as the neutrophils rapidly extrude their intracellular DNA, leading to a proteinaceous net that helps trap & kill bacteria (see below)
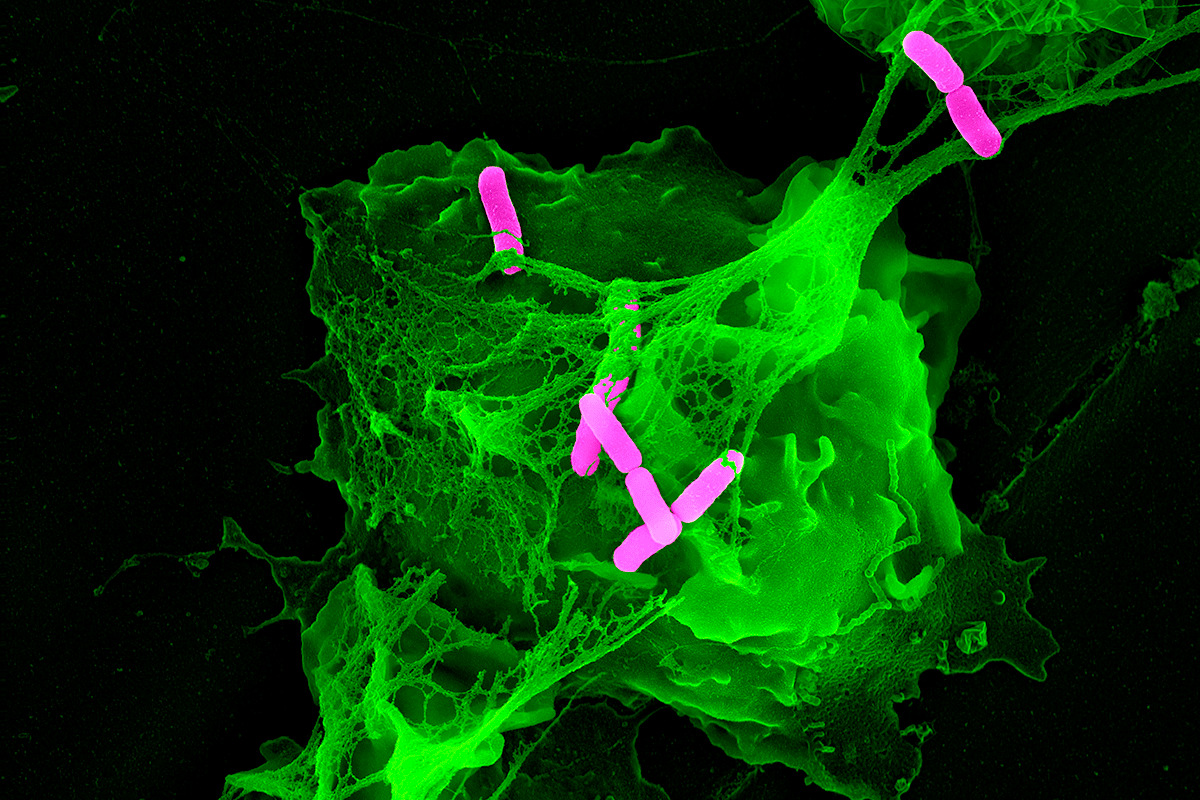
NETosis comes in two forms: “suicidal” and “vital”.
Suicidal = PMN effectively kills itself by extruding it’s nuclear DNA (green material below).
Vital = only some nuclear material exocytosed from the nucleus and released. PMN can still go on with phagocytosis afterward.
Suicidal = PMN effectively kills itself by extruding it’s nuclear DNA (green material below).
Vital = only some nuclear material exocytosed from the nucleus and released. PMN can still go on with phagocytosis afterward.
NETosis recruits red blood cells and promotes the deposition of vWF, fibrinogen, and fibrin, creating a scaffold for thrombosis.
Notably, this closely mirrors what has been seen pathologically in COVID-19!
⬆️ PMN NETs are seen, particularly in those with severe disease:
Notably, this closely mirrors what has been seen pathologically in COVID-19!
⬆️ PMN NETs are seen, particularly in those with severe disease:

Also, this is partly why fibrinogen is an acute phase reactant!
⬆️circulating fibrinogen = more scaffolding material available = better bacterial defense.
In fact, marked hyperfibrinogenemia (i.e. >700 mg/dL) ⬆️ platelet interaction, ⬆️viscosity, and confers AC resistance!
⬆️circulating fibrinogen = more scaffolding material available = better bacterial defense.
In fact, marked hyperfibrinogenemia (i.e. >700 mg/dL) ⬆️ platelet interaction, ⬆️viscosity, and confers AC resistance!
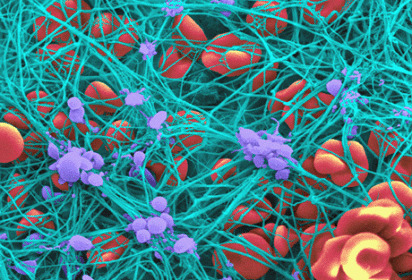
NETosis is tightly regulated by platelets.
Platelets can also release dense granules containing inorganic polyphosphate (“polyPs”), varying-length chains of negatively charged phosphate units.
PolyPs are potently procoagulant and initiate coagulation by binding FXII & thrombin.
Platelets can also release dense granules containing inorganic polyphosphate (“polyPs”), varying-length chains of negatively charged phosphate units.
PolyPs are potently procoagulant and initiate coagulation by binding FXII & thrombin.
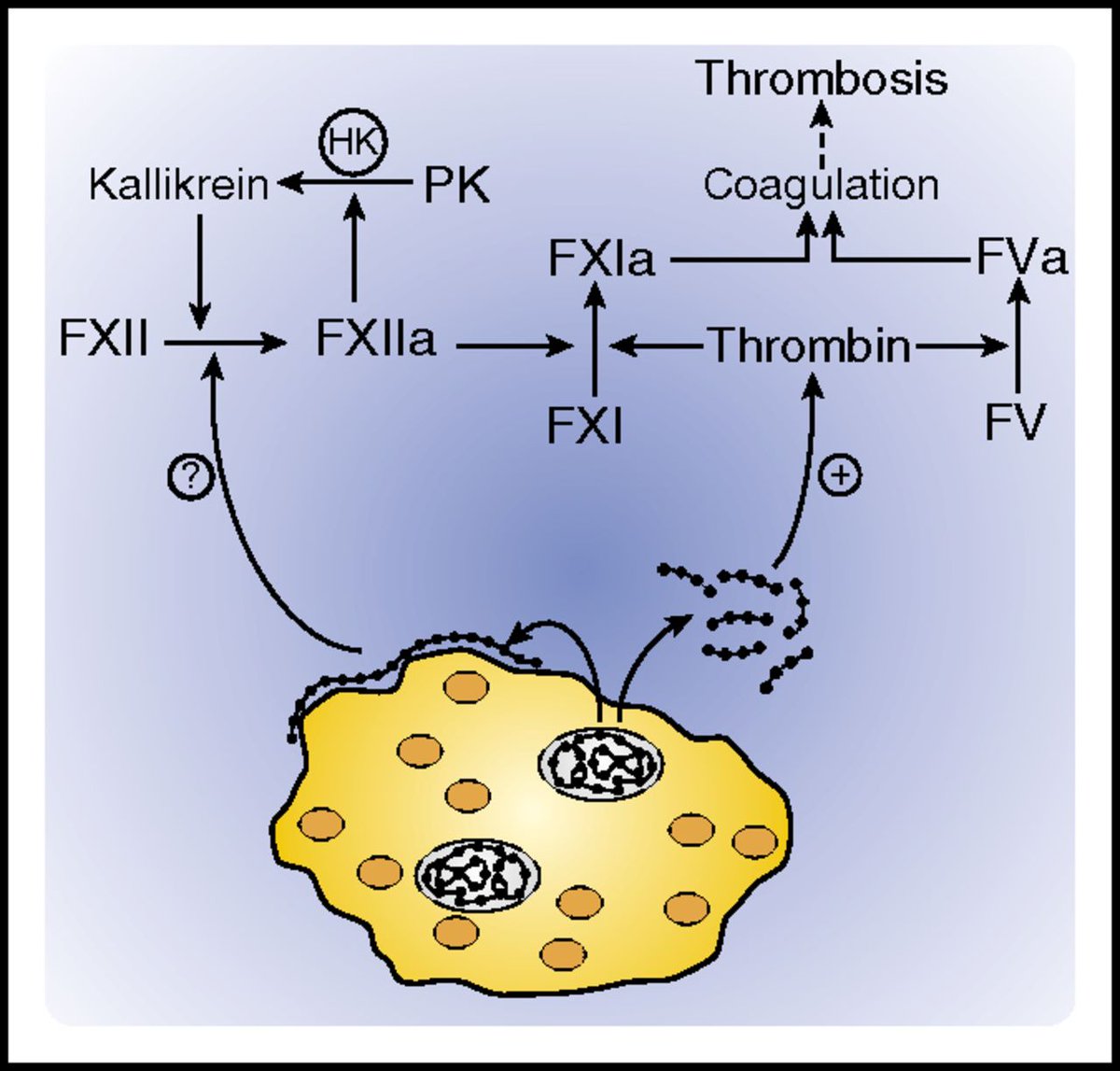
To make matters worse, we discussed in an earlier tweetorial about how vascular endothelial ACE2 expression may lead to⬆️viral invasion and accelerate vascular endothelial injury.
Since that time, endotheliitis from SARS CoV2 infection has been pathologically shown (Varga et al)
Since that time, endotheliitis from SARS CoV2 infection has been pathologically shown (Varga et al)

All of this intravascular immune activity, complement activation, when coupled with direct vascular endothelial invasion, ultimately leads to significant endothelial injury.
How would vascular endothelial injury promote thrombosis?
How would vascular endothelial injury promote thrombosis?
All of the above would lead to further thrombosis.
TEG studies from Italy have demonstrated severe platelet hyperactivation and subsequent hypercoagulability.
Plasminogen activator-inhibitor (PAI-1) is released from damaged endothelium and leads to local hypofibrinolysis.
TEG studies from Italy have demonstrated severe platelet hyperactivation and subsequent hypercoagulability.
Plasminogen activator-inhibitor (PAI-1) is released from damaged endothelium and leads to local hypofibrinolysis.
Interestingly, despite endothelial damage, bleeding is rare in COVID-19, suggesting the presence of a prothrombotic > hemorrhagic state.
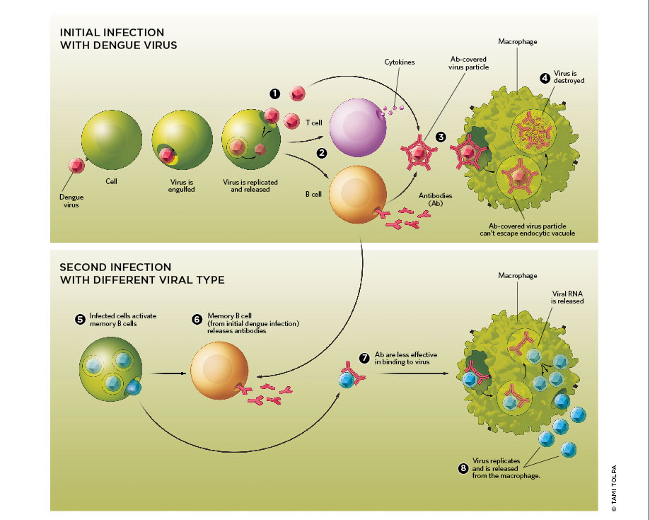
This is in stark contrast to Dengue fever, where hemorrhagic shock is the defining feature of severe disease.
...so why are they different?
This is in stark contrast to Dengue fever, where hemorrhagic shock is the defining feature of severe disease.
...so why are they different?
Remember the cells each virus infects:
Dengue principally infects macrophages → cytokine release → mast cell degranulation → vascular endothelium intact but functional → capillary leak → hemorrhagic shock
Dengue principally infects macrophages → cytokine release → mast cell degranulation → vascular endothelium intact but functional → capillary leak → hemorrhagic shock

SARS CoV2 → ACE-2 receptor → numerous cell types invaded, including vascular endothelium → complement activation / immune activation→ endothelial damage → predominantly prothrombotic phenotype
So, to summarize, what is the primary mechanism by which thrombosis occurs in COVID-19?
Where does thrombosis primarily first occur?
And why does thrombosis occur?
TL;DR:
1. COVID coagulopathy is multifactorial, but mainly represents thromboinflammation, not overt DIC.
2. PMN NETs + platelet activation + direct viral endothelial invasion + complement activation = endothelial damage → ⬆️TF and PAI-1 release→ initial pulmonary LIC
1. COVID coagulopathy is multifactorial, but mainly represents thromboinflammation, not overt DIC.
2. PMN NETs + platelet activation + direct viral endothelial invasion + complement activation = endothelial damage → ⬆️TF and PAI-1 release→ initial pulmonary LIC
3. Further embolization and/or systemic endothelial damage = markedly increased DVT/VTE risk
4. Thromboinflammation mirrors what is seen pathologically in COVID-19, with increased PMN NETs, complement deposition, endotheliitis with capillary damage, and microvascular thrombosis
4. Thromboinflammation mirrors what is seen pathologically in COVID-19, with increased PMN NETs, complement deposition, endotheliitis with capillary damage, and microvascular thrombosis
If you have read this far, thank you so much.
I know this is a controversial topic and I am always open to further data/suggestions.
In my last tweetorial, we will attempt to tie everything together, providing a framework to understand potential treatment options.
I know this is a controversial topic and I am always open to further data/suggestions.
In my last tweetorial, we will attempt to tie everything together, providing a framework to understand potential treatment options.
Further reading if interested:
Amazing tweetorial from @acweyand on data surrounding this:
Best commentary on COVID-coagulopathy to date IMO:
onlinelibrary.wiley.com/doi/epdf/10.11…
Immunothrombosis:
onlinejacc.org/content/early/…
Endotheliitis:
thelancet.com/journals/lance…
Amazing tweetorial from @acweyand on data surrounding this:
Best commentary on COVID-coagulopathy to date IMO:
onlinelibrary.wiley.com/doi/epdf/10.11…
Immunothrombosis:
onlinejacc.org/content/early/…
Endotheliitis:
thelancet.com/journals/lance…
Pathology:
medrxiv.org/content/10.110…
ncbi.nlm.nih.gov/pmc/articles/P…
medrxiv.org/content/10.110…
PMN NETs:
haematologica.org/content/haemat…
ADE SARS
sciencedirect.com/science/articl…
Polyphosphates:
ncbi.nlm.nih.gov/pubmed/22517894
medrxiv.org/content/10.110…
ncbi.nlm.nih.gov/pmc/articles/P…
medrxiv.org/content/10.110…
PMN NETs:
haematologica.org/content/haemat…
ADE SARS
sciencedirect.com/science/articl…
Polyphosphates:
ncbi.nlm.nih.gov/pubmed/22517894