Piecing together what we know of the ‘new #variant’. This has been confirmed by COG-UK (tinyurl.com/ybfsqurf) and PHE (tinyurl.com/yacjerx6) as a lineage of #SARSCoV2 carrying a set of mutations including #N501Y, found in the #spike receptor binding domain.
#PHE reports suggest that #SARSCoV2 carrying this variant(s) have been observed at high frequency in recent times in the South of England. At the moment there is no evidence this #variant spreads any faster or any slower. This simply hasn’t been tested yet (and of course will be)
This variant is found in an expanding outbreak but may be going up in frequency just by chance – we always have to disentangle these forces. Viruses mutate (actually #SARSCoV2 a bit slower than many viruses) and the presence of mutations in of themselves is not a cause for alarm.
N501Y was first observed back in April (#CoVGLUE) in a Brazilian isolate & in Australia over June. In recent times it’s been observed in S Africa, UK and DNK. This mutation has been shown to increase binding affinity in work from @jbloom_lab (jbloomlab.github.io/SARS-CoV-2-RBD…)...
...it is potentially adaptive in mice (tinyurl.com/y8bgnq4x). We pick up a different variant at this position as putatively adaptive in mink infections (tinyurl.com/y9mdxl2l).
N501Y has also been found in association with spike #deletions 69/70. del69/70 appears in distinct lineages (below current to 30/11/20) & can associate with binding domain #spike mutations, including N439K also suggested to play a role in antibody escape tinyurl.com/y3ayjkel. 
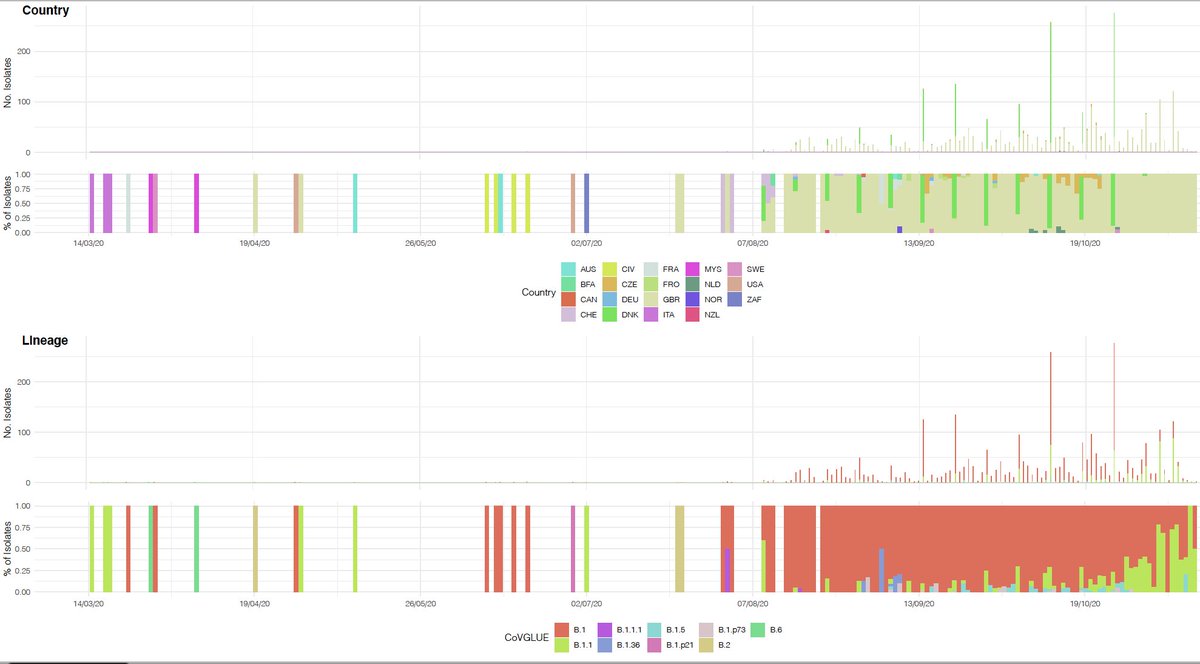
Though deletions in this region of #spike have existed in sequenced isolates since mid March in human isolates, and predate the Danish mink outbreaks.
This preprint just out from @GuptaR_lab picks up some further candidates biorxiv.org/content/10.110… and formerly reports the recurrent emergence of the #spike double deletion.
It’s clear we need to be looking for #indels in #SARSCoV2 not just replacement events.
It’s clear we need to be looking for #indels in #SARSCoV2 not just replacement events.
Given the existence of at least some of these mutations during the early 1st N hemisphere wave, it is likely all a bit more complicated than one mutation or deletion having an enormous effect on any of vaccine effectiveness, antibody recognition, disease severity or transmission.
But we should remain vigilant. We want to pick up ‘potentially’ concerning mutations early, as in this case. This is greatly aided by large genomic surveillance (@GISAID @CovidGenomicsUK @nextstrain), many data providers & the encouragement of researchers to use these resources.
At the same time there are many thousands of variants in #SARSCoV2 and the vast majority of these are expected to have no impact on #transmission (tinyurl.com/y3neo6vj). We couldn't find any in the first ~50K genomes from the #pandemic.
• • •
Missing some Tweet in this thread? You can try to
force a refresh