
,
28 tweets,
15 min read
Read on Twitter
Our paper: "Ultra-deep, long-read nanopore sequencing of mock microbial community standards", is out today! Featuring some seriously juicy @nanopore sequencing from @Scalene, and some cool assembly and analyses from me and @pathogenomenick.
academic.oup.com/gigascience/ar…
academic.oup.com/gigascience/ar…
We had a Q&A about our work with editor @SCEdmunds, talking mock metagenomes and breaking the first rule of @longreadclub gigasciencejournal.com/blog/mock-meta…
And now, a thread:
And now, a thread:
The goal of this work was to make available a really deep, long-read dataset for some community standards. We used two synthetic “mock” microbial communities manufactured by @ZymoResearch, containing a mix of gram-positive and gram-negative bacteria, and two yeasts. 
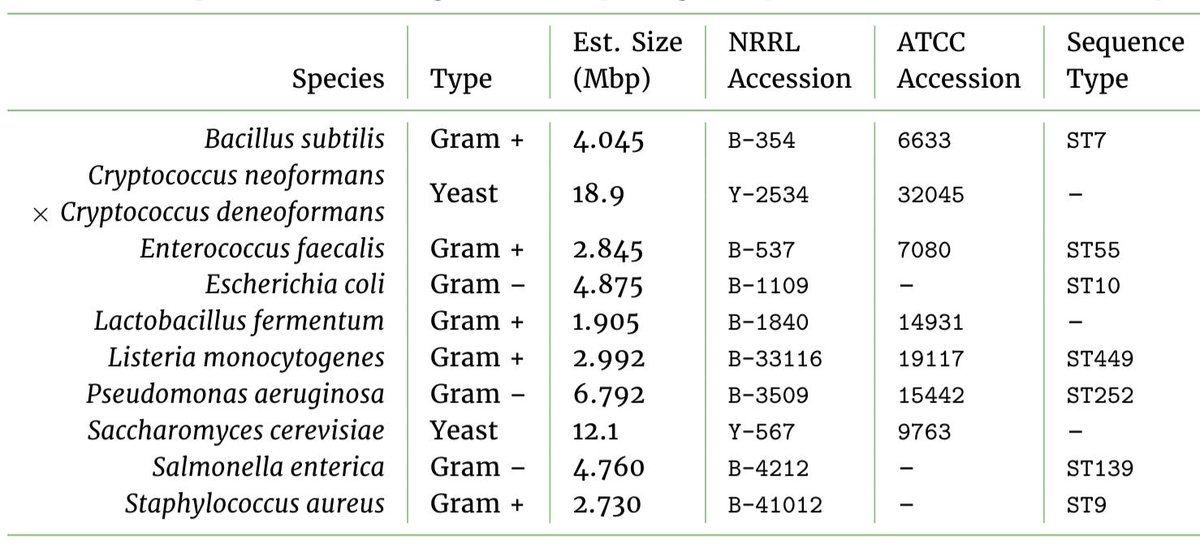
Mock communities like these are really useful for both the lab and bioinformatics as positive controls. As the samples are validated to contain a known distribution of the taxa, they could be used to check that DNA extractions, or say, taxonomic classifiers have worked properly.
As we begin to make big decisions with genomic data, it is vital that experiments are conducted correctly and that sequencing and bioinformatics pipelines can be validated. Yet, there is a lack of available testing data, particularly for metagenomics and especially long-reads.
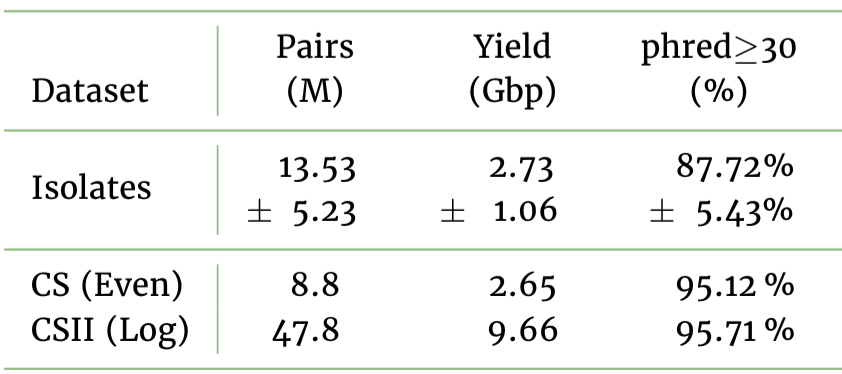
We release 14 & 16 Gbp of sequence from the GridION; with 150 & 153 Gbp from the beastly PromethION, for the even and log distributed standards respectively. Read N50 ranged 5.3 - 5.4 Kbp over the four runs. 4.2 TB of signal is available to anyone who wants to take a cluster down

Additionally, thanks to @ZymoResearch we publish Illumina sequencing runs for each of the 10 isolates, which we assembled individually into draft assemblies. We aligned our long-read nanopore metagenomics reads to the drafts to estimate taxon abundance without a database.
Aligning our @nanopore metagenomic reads to the Illumina-derived isolate references, we observed all 10 microbial species in the community were sequenced in the expected proportion. Cool: long-read metagenomics can be representative of the underlying community.
This is particularly interesting to see in the log community where species are distributed on a log scale, from 89.1% down to 0.000089% relative abundance. We could detect the lowest abundance organism using 4 GridION reads, from just 50 cells of input. Highly sensitive!

As @scalene split the sequencing libraries for the two communities between the GridION and PromethION we could directly compare the platforms. My review: it's like having a more powerful microscope. We get 10-fold more data from the PromethION at very similar quality to GridION.
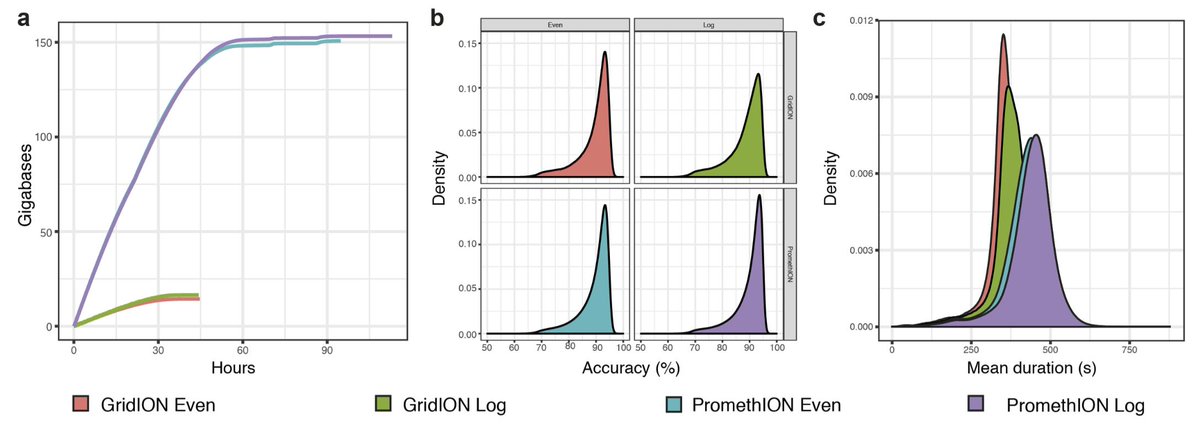
This permits us to assemble some of the lower abundance organisms from the log-distributed community and detect organisms at extremely low abundance with more confidence. This is crucial for applications where important community members are not present in the highest abundance.
We did notice a difference in sequencing speed between the PromethION (mean 419 bp/s and 437 bp/s for even and log) and the GridION (mean speed 352 bp/s and 372 bp/s) which we attributed to different running temperatures on the instrument (39C and 34C respectively).
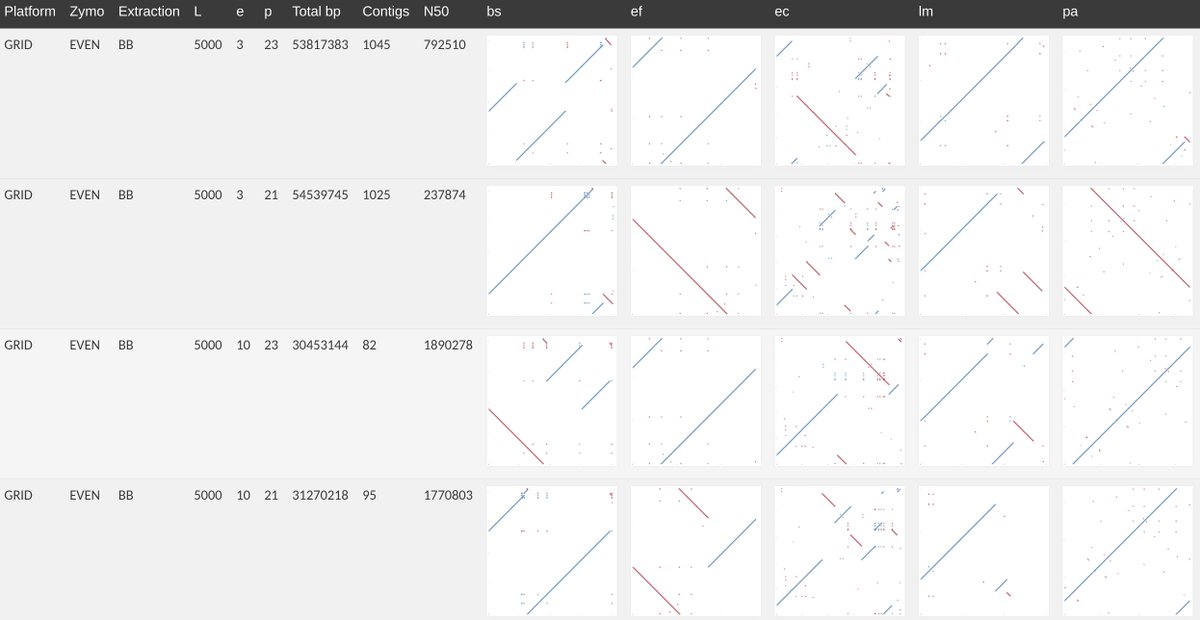
You’re probably wondering, did it assemble? I used wtdbg2 (github.com/ruanjue/wtdbg2) to construct metagenomic assemblies of both even & log communities, for GridION and a randomly subsampled 25% PromethION. It was fast, reliable and worked well. But! Parameter choices need care.
I made assemblies for log/even from Grid/Prom; varying HPC k-mer size (p), minimum edge weight (e) and read length threshold (L). However, with wtdbg2 under such active development, I gave up trying to find a parameter set that could optimise assembly size & contiguity.
Even so, for the even community; genomes near the expected size could be reliably assembled for the 8 bacteria for a variety of parameter conditions. Yeasts proved much harder to assemble but this is expected given their lower coverage. Big contigs from metagenomes can be done!
Unsurprisingly, given the log-distribution of coverage, assembling the log community posed more difficulty. Notably, Listeria the highest abundance organism (~37000× coverage) was v.poorly assembled. Very high cov seems to be a problem for the version of wtdbg2 that we used...
Great, so we have contigs, but how can we convince you they aren’t garbage? Luckily for us, Alexa McIntyre from the @mason_lab had recently published some PacBio isolate sequencing and assemblies for eight of the ten organisms in the Zymo communities nature.com/articles/s4146….
Alexa and @mason_lab were kind enough to share data ahead of their publication (thanks!), allowing us to perform a robust comparison of our nanopore assemblies. Aligning with minimap2 (-x asm10) makes for some pretty dotplots. HT @ZaminIqbal for the suggestion after our preprint.
Identities are good, but what about completeness? As suggested by review, I estimated completeness of some assemblies with checkM, comparing completeness scores after each stage of our polishing pipeline (raconx2, medaka, pilonx2). All done automatically by our snakemake pipeline
Each improves completeness. racon & medaka long-read more so (~10%), but Illumina end-polishing useful for final 1-2%. Interesting as it shows we can achieve most of what we need from polishing with just the long-reads alone.

So, yes! We can assemble genomes, with high contiguity, identity and completeness. We are getting close to single-contig finished-quality genomes without isolates, without binning, without databases. Who said long-read @nanopore metagenomics wasn’t possible huh?
Of course, it goes without saying that this data mountain wouldn't be possible without the legendary @Scalene working the pipettes to make sure our extraction protocol would be representative. The protocol is open, and you can read it here: dx.doi.org/10.17504/proto…
We've taken care to make this is a nice demonstration of what can be achieved with the cutting edge of long-read metagenomics (newest basecaller, assembler and polishing strategy), but of course, this is just the beginning and there will be plenty more to come!
We are really hoping that you will take parts (or all!) of our data and use it to develop and validate new laboratory and software pipelines for long-read metagenomics.
In the spirit of @longreadclub; our reads, software and analyses are open access and open source, because we want everybody to be able to achieve really very long reads indeed. Head to Github github.com/LomanLab/mockc… and watch our website for updates lomanlab.github.io/mockcommunity/
We've been working on this since the paper was reviewed, revised and accepted, so stay tuned over the next few days for some new data that isn't in this paper, some thoughts on @fenderglass' awesome Flye assembler and where we think we'll be going next.

Finally some thanks are owed to @jaredtsimpson, @mattloose and @DrT1973 for useful @nanopore discussions and advice. Alexa and @mason_lab for making their PacBio data available early to us, and @watermicrobe and @ljcoin for the constructive and fair reviews!
Additionally, I’d like to thank Radoslaw Poplawsk for helping me saturate cores on @MRCClimb. Also thanks to Divya Mirrington (@nanopore) for advice on taming the PromethION; and Hannah McDonnell at Cambridge Bioscience for providing the @ZymoResearch standards.














