
,
15 tweets,
4 min read
Read on Twitter
Can we please talk about this problematic manuscript?
"Very rare pathogenic genetic variants detected by SNP-chips are usually false positives: implications for direct-to-consumer genetic testing"
biorxiv.org/content/10.110…
"Very rare pathogenic genetic variants detected by SNP-chips are usually false positives: implications for direct-to-consumer genetic testing"
biorxiv.org/content/10.110…
Indeed, there are accurate and new findings in this manuscript. The problem is that the accurate findings are not new and the new findings are not accurate.
Let's start...
👇👇👇
Let's start...
👇👇👇
In a nutshell, the authors compared array results from the UK Biobank to sequencing results and could not replicate many rare positive calls. This led them to caution against DTC genomics that use arrays to test monogenic conditions with ultra rare alleles.
So what's the issue?
So what's the issue?
The accurate finding is that rare variants have low PPV. This is quite obvious since PPV≈c/(c+e), where c is the carrier rate and e is the accuracy of the probe. It is obvious and we are all aware of that. In fact, it is taught in undergraduate statistics. 
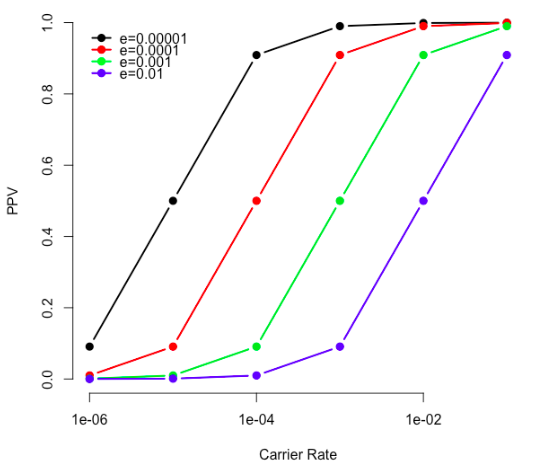
The "novelty" of the manuscript is the claim that this issue affects DTC genomics. This is a strong claim because the manuscript did not check DTC genomics pipelines at all. They simply looked at arrays in an irrelevant project and rushed to the conclusion below:

This is a lazy approach b/c if they actually wanted to test DTC companies, they could submit real positive samples rather than analyzing the UK biobank. Just because UK biobank uses arrays and DTC genomics use arrays does not mean that you compare apples-to-apples
We are well aware to the issue of low PPV with ultra rare variants and since we try our best to not be total idiots, we use a different pipeline (1) we sanger every positive rare variant in our health report (2) we have multiple probes on the array for most medical-variants ...
For instance, we have more than 5 probes for \delta F508. This improves the SNR considerably and therefore increases the PPV even without Sanger (3) we know that not all probes are created equal. In fact, the error rate show bi-modal distribution with most are highly accurate
and a minority of probes are really bad. So when we select SNPs to be in our reports, we exclude the bad probes. The authors of the study simply lumped all of the variants in the same MAF bin together (see below). This can really affect the average PPV since bad probes are there

We contacted the authors and asked for the underlying data of this figure. We did not ask for genotypes just a simple table with variant and its observed PPV to test the hypothesis that bi-modality inflates the PPV. They decline to share with us this table😱😱😱. How lovely!
Finally, we note that the authors tested arrays technologies that are irrelevant for DTC. All major companies use Illumina. They tested two arrays: BiLEVE and Axiom that use a different chemistry. Would you test error rates with ONT and project these rates to HiSeq?
Why the exact array technology is so important? Because even the authors reported substantial differences between these arrays in their study. See how the PPV changes in their Table 1 between two array technologies within the same project.

Illumina is a third technology. Maybe the PPV is totally different?
So let's summarise, we validate every positive using Sanger, our array combines multiple probes, we do not test SNPs that are likely to be erroneous, and we use a different array technology.
So let's summarise, we validate every positive using Sanger, our array combines multiple probes, we do not test SNPs that are likely to be erroneous, and we use a different array technology.
DTC genomics deserve the scrutiny of the community and we welcome feedback. But critique is only valuable when it is based on facts rather than shortcuts. Thanks for bearing with me!
I would like to point out the @carolinefwright and her team shared with us the data regarding PPV of BRCA variants since I tweeted earlier. We appreciate their efforts and commitment to share raw data.








