,
18 tweets,
4 min read
Read on Twitter
1/
Why does acute tubular necrosis decrease GFR?
There is clearly “acute kidney injury” (the tubules are damaged), but the glomerulus is intact. Why should glomerular filtration decrease if the filtering apparatus is unaffected?
Follow this thread to find out!
Why does acute tubular necrosis decrease GFR?
There is clearly “acute kidney injury” (the tubules are damaged), but the glomerulus is intact. Why should glomerular filtration decrease if the filtering apparatus is unaffected?
Follow this thread to find out!
2/
Patients with ATN experience a profound decrease in GFR (as evidenced by an increase in creatinine +/- a decrease in urine output).
Which of the following is a major contributor?
Patients with ATN experience a profound decrease in GFR (as evidenced by an increase in creatinine +/- a decrease in urine output).
Which of the following is a major contributor?
3/
Distal sodium delivery is a key factor in ATN. As part of tubular injury, the Na/K-ATPase moves from the basolateral to apical membrane (so cool!). Without the ATPase, sodium reabsorption is limited.
The unabsorbed sodium remains in the lumen.
link.springer.com/article/10.100…
Distal sodium delivery is a key factor in ATN. As part of tubular injury, the Na/K-ATPase moves from the basolateral to apical membrane (so cool!). Without the ATPase, sodium reabsorption is limited.
The unabsorbed sodium remains in the lumen.
link.springer.com/article/10.100…

4/
We make use of the decreased sodium reabsorption diagnostically as much of it ends up in the urine, increasing the fractional excretion of sodium (FeNa). An elevated FeNa is seen in ATN… this is why!
There are also pathophysiologic consequences of all this luminal sodium.
We make use of the decreased sodium reabsorption diagnostically as much of it ends up in the urine, increasing the fractional excretion of sodium (FeNa). An elevated FeNa is seen in ATN… this is why!
There are also pathophysiologic consequences of all this luminal sodium.
5/
It starts with delivery to the distal convoluted tubule. Recall what sits there: the macula densa.
And what does the macula densa do when it “sees” more sodium?
It starts with delivery to the distal convoluted tubule. Recall what sits there: the macula densa.
And what does the macula densa do when it “sees” more sodium?
6/
Given that the distal sodium is seen by the macula densa as evidence of increased GFR, the prudent response would be to vasoconstrict the afferent arteriole, decreasing hydrostatic pressure in the glomerulus.
This leads, finally, to decreased GFR!
Given that the distal sodium is seen by the macula densa as evidence of increased GFR, the prudent response would be to vasoconstrict the afferent arteriole, decreasing hydrostatic pressure in the glomerulus.
This leads, finally, to decreased GFR!
7/
But, afferent vasoconstriction is a normal response to increased delivery of sodium distally (“tubuloglomerular feedback”).
If this were enough to cause a dramatic fall in GFR, we’d see it all the time after thiazide diuretic administration. Something else must be going on.
But, afferent vasoconstriction is a normal response to increased delivery of sodium distally (“tubuloglomerular feedback”).
If this were enough to cause a dramatic fall in GFR, we’d see it all the time after thiazide diuretic administration. Something else must be going on.
8/
The afferent vasoconstriction response appears to be exaggerated and sustained in ischemic injury.
The cause is multifactorial (with a big component being the inflammatory response to ischemia), but the key point is that vasoconstriction continues, limiting renal blood flow.
The afferent vasoconstriction response appears to be exaggerated and sustained in ischemic injury.
The cause is multifactorial (with a big component being the inflammatory response to ischemia), but the key point is that vasoconstriction continues, limiting renal blood flow.
9/
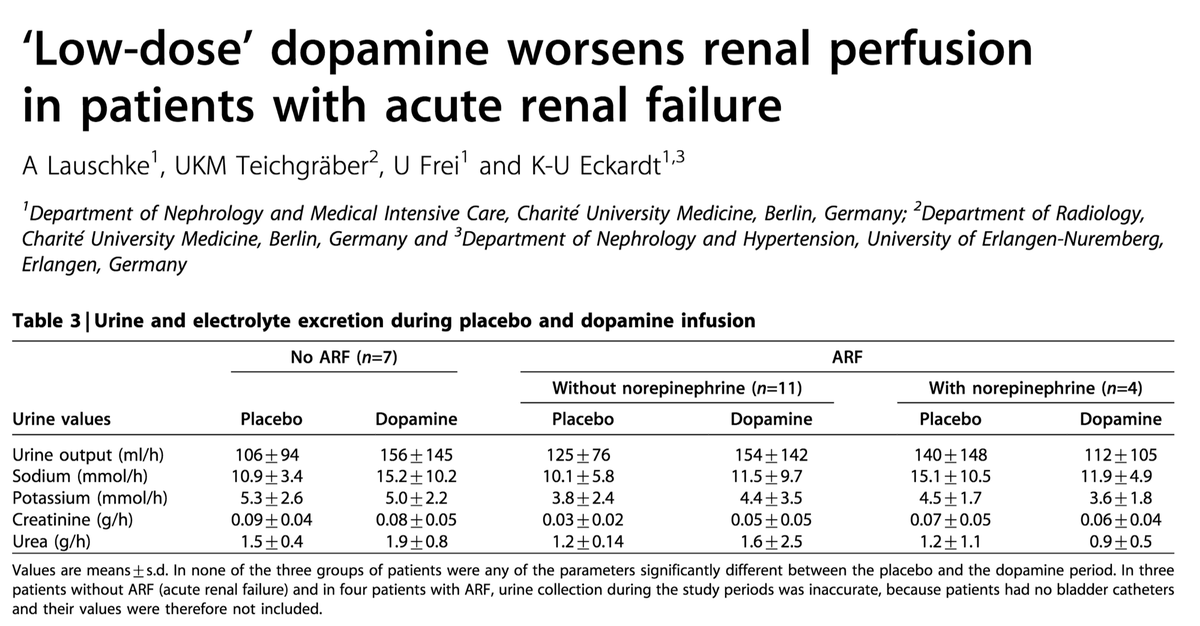
Understanding the above, makes it clear why so many trials of dopamine to prevent/treat ATN have been attempted. If we can just vasodilate the renal arterioles (“renal does” dopamine can do this!), maybe we can stop the main pathology of ATN.
Understanding the above, makes it clear why so many trials of dopamine to prevent/treat ATN have been attempted. If we can just vasodilate the renal arterioles (“renal does” dopamine can do this!), maybe we can stop the main pathology of ATN.
10/
Unfortunately, all the factors in #8 above suggest that the renal vascular is insensitive to usual vasodilatory factors.
Result: negative trials.
e.g.: linkinghub.elsevier.com/retrieve/pii/S…
Unfortunately, all the factors in #8 above suggest that the renal vascular is insensitive to usual vasodilatory factors.
Result: negative trials.
e.g.: linkinghub.elsevier.com/retrieve/pii/S…
11/
(At least) two mysteries remain:
For the first, I’ll provide an answer.
For the second, I'll call on Twitter for explanations.
(At least) two mysteries remain:
For the first, I’ll provide an answer.
For the second, I'll call on Twitter for explanations.
12/
Mystery #1: If distal sodium delivery is a key driver of ATN, why have so many trials of furosemide for the treatment of ATN been done? There are many explanations (e.g., flushing of tubular debris), but the one that I find coolest relates to furosemide's mechanism of action.
Mystery #1: If distal sodium delivery is a key driver of ATN, why have so many trials of furosemide for the treatment of ATN been done? There are many explanations (e.g., flushing of tubular debris), but the one that I find coolest relates to furosemide's mechanism of action.
13/
We all know furosemide as a “loop diuretic”. In the loop of Henle, furosemide inhibits the Na-K-Cl co-transporter (specially NKCC2).
Here’s the key: NKCC2 resides someplace else in the kidney…
We all know furosemide as a “loop diuretic”. In the loop of Henle, furosemide inhibits the Na-K-Cl co-transporter (specially NKCC2).
Here’s the key: NKCC2 resides someplace else in the kidney…
14/
...in the macula densa!
Furosemide blocks tubuloglomerular feedfback (see #6 and #7 above) by blocking the macula densa from “seeing” the increase distal sodium. How cool is that?
jci.org/articles/view/…
...in the macula densa!
Furosemide blocks tubuloglomerular feedfback (see #6 and #7 above) by blocking the macula densa from “seeing” the increase distal sodium. How cool is that?
jci.org/articles/view/…

15/
Mystery #2: If distal sodium delivery is important to the mechanism of ATN, why aren’t these patients polyric instead of oliguric?
I have a hypothesis, but it's unconfirmed and I'll hold it for now.
#nephJC — what’s your explanation?
Mystery #2: If distal sodium delivery is important to the mechanism of ATN, why aren’t these patients polyric instead of oliguric?
I have a hypothesis, but it's unconfirmed and I'll hold it for now.
#nephJC — what’s your explanation?
16/
There are other mechanisms at play in ATN. I’ll mention one other: tubular obstruction.
There are other mechanisms at play in ATN. I’ll mention one other: tubular obstruction.
17/
After ischemic injury, the tubules fill with cellular debris. This causes obstruction, which might lead to:
- increased tubular pressures
- increased pressures in Bowman’s space
- decreased flow across the glomerular capillary
- decreased GFR
After ischemic injury, the tubules fill with cellular debris. This causes obstruction, which might lead to:
- increased tubular pressures
- increased pressures in Bowman’s space
- decreased flow across the glomerular capillary
- decreased GFR

18/
Let’s return to the original question:
Patients with ATN experience a profound decrease in GFR. Which of the following is a major contributor?
Let’s return to the original question:
Patients with ATN experience a profound decrease in GFR. Which of the following is a major contributor?