Bon vue que c'est bientôt la fin de semaine, on reprend le thread sur mon sujet !!! #thèsolie
#thread #bioinformatique #assemblage
#thread #bioinformatique #assemblage
Je vous avez laissé avec quelque notions important, que je vais rappelé rapidement :
- Le séquençage c'est ce qui permet de lire la séquence ADN, mais on ne peut lire que des petits morceaux et on sais pas ou on lis du coup on est un peut embêté pour comprendre ce qu'on a lue
- Le séquençage c'est ce qui permet de lire la séquence ADN, mais on ne peut lire que des petits morceaux et on sais pas ou on lis du coup on est un peut embêté pour comprendre ce qu'on a lue
- L'assemblage c'est l'étape algorithmique qui vas permettre de reconstruire la séquence d'ADN d'origine a partir des morceaux qu'on a lue. On fais sa parce que les morceaux partage des séquences communes, sa s'appel des overlaps. Pour sa on utilise un graph d'assemblage
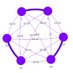
- Dans le graphes d'assemblage on représente les morceaux d'ADN qu'on a lue sous forme de nœud, et quand il partage de séquence, qu'il s'overlap on les relis par des arc. Souvenez vous: 

- Les répétition: le graphe est simple mais il continent une #exempleBienConstruit Une répétition c'est un bout d'ADN qu'on retrouve a plusieurs fois dans la séquence entière, et elle pose problème.
Quand dans le graphe on arrive dans une répétition on sais pas par ou sortir.
Quand dans le graphe on arrive dans une répétition on sais pas par ou sortir.
- Les contigs: vue qu'on ne sais pas résoudre ce problème les algorithme on crée des contigs. Un contig c'est un bout du graph ou il n'y a pas de répétition.
Bon je crois qu'on a fini les rappelles, maintenant on vas parler de quelque chose que j'ai parler très rapidement les technologies de séquençages.
Il existe trois générations de séquençage :
- La première la technique Sanger du nom du créateur du séquençage Frederick Sanger il est mignon non ? Moi je le trouve gentil
- La première la technique Sanger du nom du créateur du séquençage Frederick Sanger il est mignon non ? Moi je le trouve gentil

Je vais pas vous expliquer comment le séquençage Sanger marche ce qu'il faut retenir c'est que sa coute très chère sa prend beaucoup de temps on fais des morceaux petit (quelque centaine de bases), mais c'est très très précis cette technique fais très très rarement d'erreur.
- La second génération:
On fais des fragments encore plus petit qu'avec la technique Sanger (45 à 400 bases), on fais une erreur tout les 100 a 1000 bases. Mais c'est beaucoup plus rapide, sa coute beaucoup moins chère. Du coup tout le monde en a fais.
On fais des fragments encore plus petit qu'avec la technique Sanger (45 à 400 bases), on fais une erreur tout les 100 a 1000 bases. Mais c'est beaucoup plus rapide, sa coute beaucoup moins chère. Du coup tout le monde en a fais.
- La 3ème générations:
On fais des fragments bcp bcp + long (jusqu'a 60.000 bases), mais sa coute un plus chère mais le vrais pb c'est qu'on fais plus d'erreur entre 15 % et 30 %
Imaginé vous entrain de lire un livre et de ce trompé 15 fois tout les 100 lettres.
On fais des fragments bcp bcp + long (jusqu'a 60.000 bases), mais sa coute un plus chère mais le vrais pb c'est qu'on fais plus d'erreur entre 15 % et 30 %
Imaginé vous entrain de lire un livre et de ce trompé 15 fois tout les 100 lettres.
Si ces technologies ce trompe si souvent pourquoi on les utilises et bien a cause des répétitions dans l'exemple qui on une taille de 3 mais si on avait des lectures de taille 5 et bien on pourrais résoudre l'assemblage. Le graphe d'assemblage serais linéaire.

On peut dire que si les reads sont plus long que les répétitions on peut résoudre l'assemblage. Une autre image pour appuyé l'idée.
En vert, rouge et bleu sur les cotés du graphes des "reads" si on suit les chemins que ces reads nous indique on peut traversé la répétition.
En vert, rouge et bleu sur les cotés du graphes des "reads" si on suit les chemins que ces reads nous indique on peut traversé la répétition.

Du coup vous comprenez bien que quand on passe de fragment (ou reads c'est synonyme) qui on une taille de 150 base a 20.000 bases on pas pouvoir résoudre plus de répétition.
C'est ce constat qui a poussé Sergey Koren et Adam MPhillippy a écrire en 2015 un article
C'est ce constat qui a poussé Sergey Koren et Adam MPhillippy a écrire en 2015 un article
"One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly" doi.org/10.1016/j.mib.… #uneBonneThèseCiteCesSource
En gros ils dissent qu'étant donnée que les répétitions chez les bactéries sont plus petites que les reads de 3 ème génération
En gros ils dissent qu'étant donnée que les répétitions chez les bactéries sont plus petites que les reads de 3 ème génération
le problème de l'assemblage des bacteries est résolue.
Pour résumé "Les gars les biochimistes on résolue le problème, il y a quoi d'autre a faire ?"
Sauf que c'est pas tout a fais vrais il reste des cas ou sa ne marche pas.
Pour résumé "Les gars les biochimistes on résolue le problème, il y a quoi d'autre a faire ?"
Sauf que c'est pas tout a fais vrais il reste des cas ou sa ne marche pas.
Et mon sujet c'est sa :
"Pourquoi l'assemblage des génomes bactériens avec des reads de 3ème générations n'est pas résolue alors qu'il devrait !"
"C'est pas le vrais sujet il est beaucoup plus tarabiscoté que sa en vrais !" @nmarijon Part En Thèse #uneBonneThèseCiteCesSource
"Pourquoi l'assemblage des génomes bactériens avec des reads de 3ème générations n'est pas résolue alors qu'il devrait !"
"C'est pas le vrais sujet il est beaucoup plus tarabiscoté que sa en vrais !" @nmarijon Part En Thèse #uneBonneThèseCiteCesSource
Et c'est fini pour aujourd'hui on parlera de comme on peut résoudre ce problème une autre fois !
#Thésolie #bioinformatique #vulgarisation
@threadreaderapp unroll please !
#Thésolie #bioinformatique #vulgarisation
@threadreaderapp unroll please !









