,
16 tweets,
7 min read
Read on Twitter
Do you work with single cell RNA-Seq data (or any count data with large numbers of zeros)? You may be interested in our preprint now available on @biorxiv_genomic : biorxiv.org/content/10.110… in collaboration with @rafalab @stephaniehicks and Martin Aryee
I will try to describe here the main points. 1. Sampling distribution of unique molecular identifier counts (UMI) is NOT zero inflated (see also nxn.se/valent/2017/11… from @vallens ). Apparent zero inflation in read counts is due to PCR duplicates. 
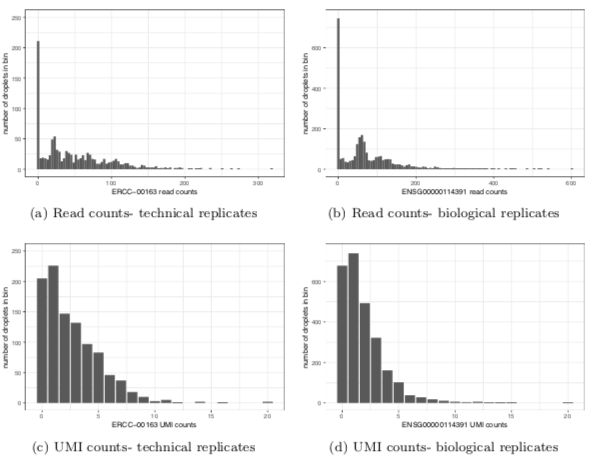
2. UMI counts are well-described by multinomial sampling ("competition to be counted") and only contain relative information (ie like microbiome they are compositional). This has been pointed out before by @inschool4life , see eg biorxiv.org/content/10.110…
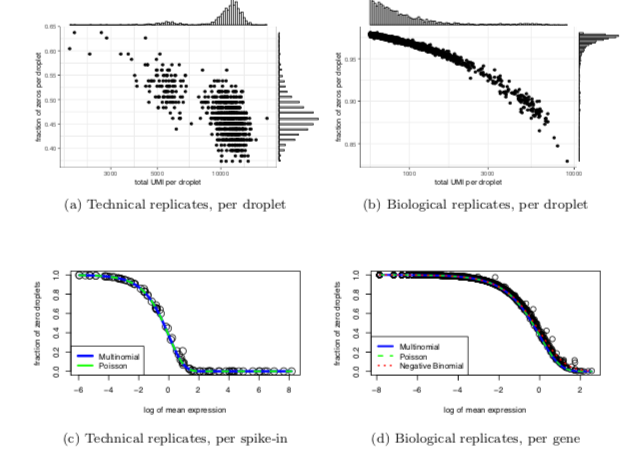
3. When the number of categories (genes) is high, and no single gene dominates all the others, the multinomial distribution can be well approximated by independent Poissons, using the total count as an offset.
4. To account for overdispersion, ideally one should use a Dirichlet-multinomial distribution. However, this can be approximated by independent negative binomial distributions.
5. Using log of counts per million (CPM) as normalization is a bad idea when most of the data are zeros. The log transform strongly distorts the difference between zero and small nonzero values based on an arbitrary pseudocount (see also Aaron Lun biorxiv.org/content/10.110… )
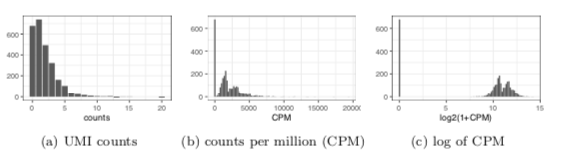
6. PCA on log-CPM recovers variation in total counts (equivalently, fraction of zeros) as the primary axis of variation even after normalization. The point of normalization is to remove this variation. The log transform effectively reverses the normalization.

7. More generally, any normalization with size factors < 1 will show similar behavior by inflating gap between zeros and nonzeros. Bottom line, we want to avoid normalization and just directly model the counts (same idea as DESeq2 and edgeR for differential expression)
8. PCA implicitly uses a Gaussian likelihood. We implement a generalized PCA (GLM-PCA) that handles Poisson, NB likelihoods as approximation to the multinomial. The code is here: github.com/willtownes/scr… . Similar idea to ZINB-WAVE (@drisso1893 ) but without zero inflation
9. GLM-PCA removes the correlation between PC1 and the fraction of zeros, total counts.

10. Approximation to GLM-PCA that is as fast as regular PCA: fit a multinomial null model of constant gene expression across cells, use the (approximate) deviance residuals as input to PCA.
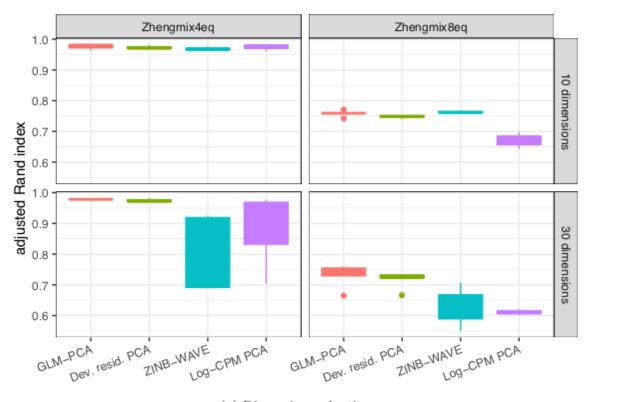
11. GLM-PCA and its fast approximation outperform PCA on log-CPM in a clustering assessment with ground truth labels (same data as f1000research.com/articles/7-114… ). ZINB-WAVE also does well but is much slower and not robust to choosing the right number of latent dimensions.
12. Most dimension reductions start by pre-filtering genes to include only top 500-2,000 "highly variable" genes. We suggest using an approximate multinomial deviance instead. This improves clustering performance even for suboptimal dimension reductions

13. Intuition behind deviance: we fit a multinomial null model of constant gene expression across cells. Deviance quantifies the "goodness of fit" of that null model. If deviance is low, probably no biological variability and gene can be discarded.
14. In conclusion, stop using log-CPM to normalize single cell RNA-Seq. If must normalize, use deviance residuals. Otherwise, avoid normalization by using direct count models (GLM-PCA for dimension reduction). Use deviance for feature selection instead of highly variable genes.
Thanks also to @keegankorthauer , @areyesq , @yered_h , Jeff Miller (jwmi.github.io), and Linglin Huang for helpful suggestions!