,
20 tweets,
5 min read
Read on Twitter
Thanks @aayushraman @tangming2005. Here's a few thoughts on how I see the landscape of tools (thread) 1/n
In HiChIP data analyses, there are two primary problems that we are trying to solve. A) Which anchors (i.e. genomic loci) should be used as a feature set and B) which loops (i.e. interactions between pairs of loci) are important in the data. 2/n
Depending on what you are hoping to use your data for, there are a variety of ways to think about anchors and loops. Two uses of HiChIP that come to mind are "which gene is this enhancer talking to" and "which loops are differential between my celltype/condition of interest" 3/n
When Martin and I wrote hichipper, we envisioned the second question being more used (i.e. building out a framework for differential loop calling), so we wanted a pre-processing pipeline that was as inclusive of potential loops as possible that could be subsetted downstream 4/n
To these ends, we reported an improved version of anchor detection from HiChIP data by modeling the restriction enzyme cut bias explicitly, which helped identify high-quality anchors from the data itself 5/n 

(we achieve this by re-parametrizing MACS2 peak calling by essentially fitting a loess curve to the data in the previous picture) 6/n

Unfortunately, based on user feedback, this modified background winds up with a very, very conservative peak calling if the library preparations are sub-par. Thus, the safest way to approach HiChIP data analyses is often to use a pre-defined anchor set 7/n
These can be from either a complementary ATAC-seq or ChIP-seq dataset for the conditions that you are interested in. From what I've seen, you can supply a bed file to hichipper or other tools directly. Hichipper does some other modifications by default to this bed file FYI 8/n
In terms of the second problem of identifying loops, hichipper didn't make any revolutionary progress. We recommend some level of CPM-based filtering + mango FDR calculation (implemented in hichipper) for identifying single-library significant loops. 9/n
Where I've personally done the most is getting multiple libraries from multiple conditions and using some sort of between-replicate logic to filter to a reasonable (~10,000-20,000) number of loops ( see e.g. github.com/caleblareau/k5…) 10/n
Other tools (that I admittedly have not tried) use a variety of statistical techniques to (probably more intelligently from what I can tell) merge anchors or filter loops for analyses. A brief run down of those that I'm aware of (not exhaustive)-- 11/n
MAPS (biorxiv.org/content/biorxi…) uses a measure of reproducibility with ChIP-seq to define a normalization and significance basis for loop calling. Given HiChIP-specific restriction enzyme bias, this seems sensible 12/n
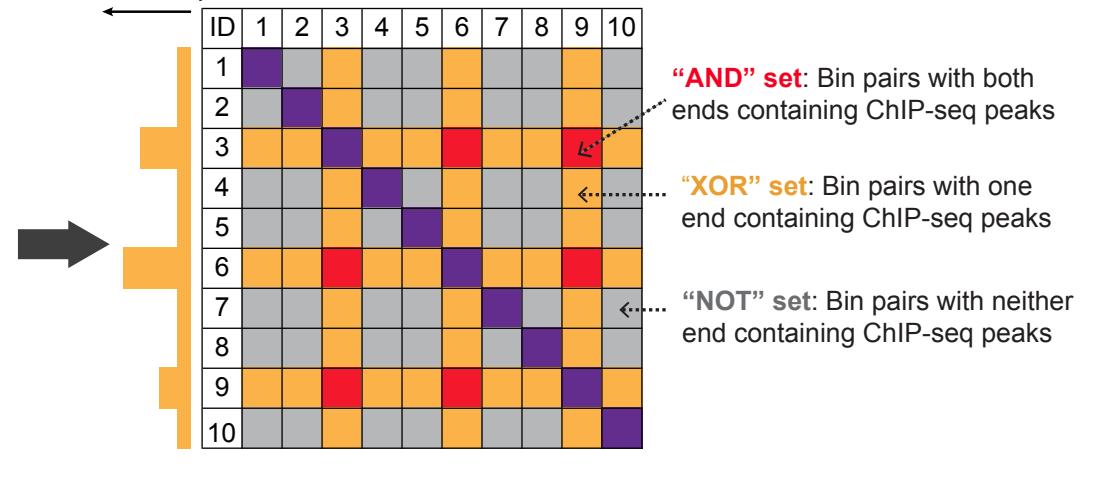
FitHiChIP (biorxiv.org/content/early/…) provides automatic merging of nearby anchors to solve the "hairball" problem, which is clearly shown Fig. 1. When I compared hichipper to FitHiC, the bias regression seemed to perform well, but I ran into memory issues which high... 13/n
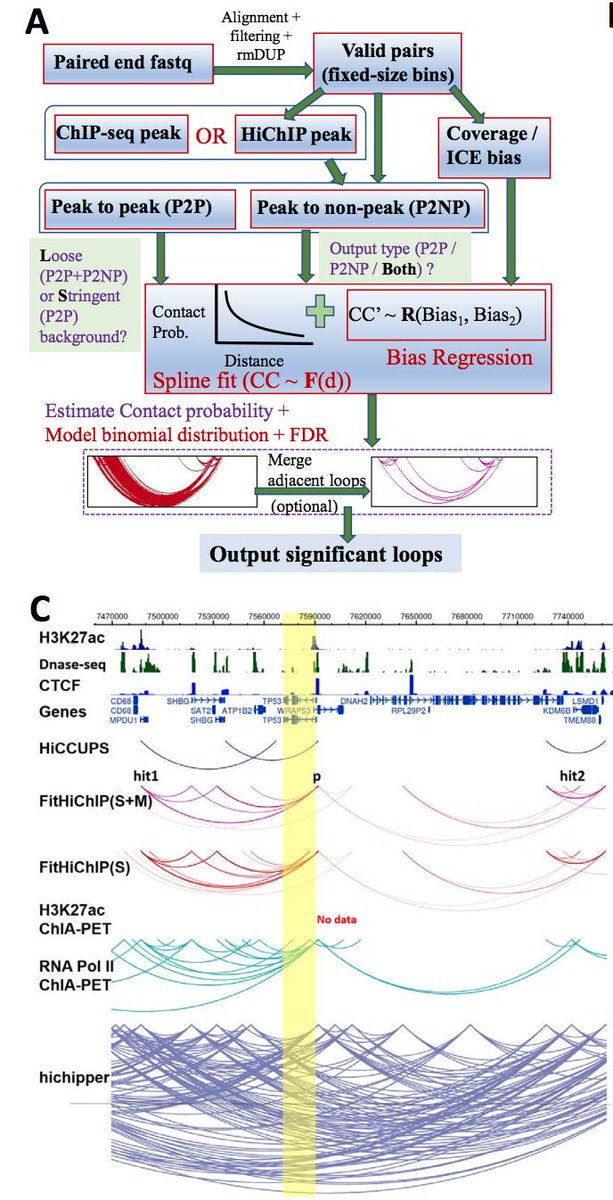
resolution (i.e. ~2.5kb) HiChIP data, which the authors have apparently solved in FitHiChIP. 14/n
Additionally, there is CID, which uses a density-based method to further collapse anchors to solve the "hairball" problem. 15/n

There are certainly other tools out there, but from my experience, any of these four (hichipper, MAPS, FitHiChIP, and CID) will probably give you something sensible (again acknowledging that I myself haven't actually run these other 3 tools) 16/n
And if you're still reading this, I'll be a bit more specific about how I view hichipper pros/cons from both my own use and others in the community: hichipper provides the most "vanilla" functionality to given sensible yet exhaustive anchors and loops. 17/n
I prefer it this way because I find that for each data set, I have to apply variable downstream threshold and cutoffs because the assay is so variable depending on which experimentalist performs the protocol and the biological question often varies so much 18/n
This may be a negative for individuals new to bioinformatics or HiChIP data but seemingly a positive for someone more experienced in working with related data. It's not obvious to me which other tools may be more applicable to a novice 19/n
Hope this helps paint a picture-- do let me know what you find if you compare tools! I think that it would be useful for the community. 20/20