,
18 tweets,
7 min read
Read on Twitter
1/
Why does acute pulmonary embolism (PE) cause hypoxia?
It took me a while to realize I didn't have a good answer. Think about it: the blood upstream of the clot should be fully oxygenated. Where is the blood that isn't being oxygenated?
The answer is pretty cool...
Why does acute pulmonary embolism (PE) cause hypoxia?
It took me a while to realize I didn't have a good answer. Think about it: the blood upstream of the clot should be fully oxygenated. Where is the blood that isn't being oxygenated?
The answer is pretty cool...

2/
Two questions to start.
First, which of the following is the major mechanism by which pulmonary embolism leads to hypoxia?
Two questions to start.
First, which of the following is the major mechanism by which pulmonary embolism leads to hypoxia?
3/
Acute PE leads to hypoxia primarily via V/Q mismatch. But, what's altered? And, in what direction?
[V=ventilation; Q=perfusion]
Acute PE leads to hypoxia primarily via V/Q mismatch. But, what's altered? And, in what direction?
[V=ventilation; Q=perfusion]
4/
While PE surely causes decreased Q, this leads to deadspace ventilation (i.e., areas of V but no Q). Deadspace ventilation does not cause hypoxia.
So, although decreased Q occurs, it's not the answer.
What is the cause? INCREASED Q! Let's see how.
ncbi.nlm.nih.gov/pubmed/25006441
While PE surely causes decreased Q, this leads to deadspace ventilation (i.e., areas of V but no Q). Deadspace ventilation does not cause hypoxia.
So, although decreased Q occurs, it's not the answer.
What is the cause? INCREASED Q! Let's see how.
ncbi.nlm.nih.gov/pubmed/25006441
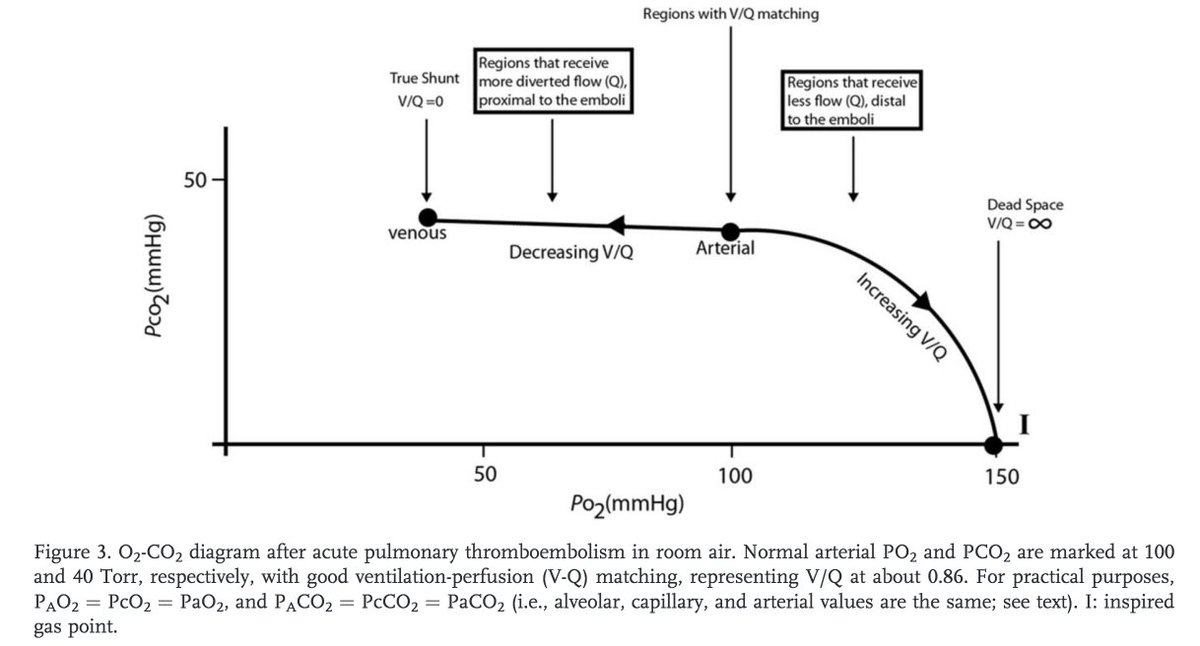
5/
Two clues that the mechanism isn't related to the thrombus itself (i.e., decreased Q):
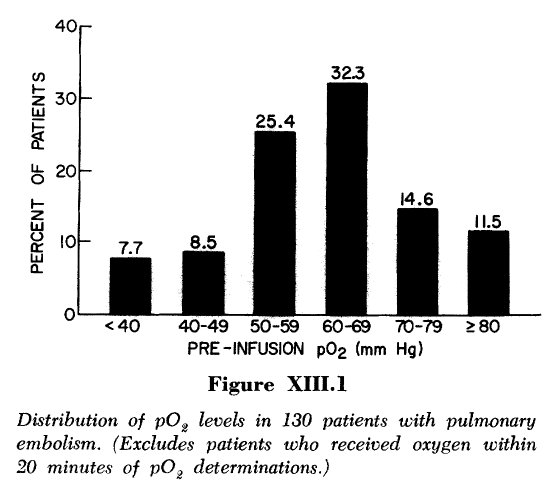
1. PaO2 varies widely, even in those with large PEs (Pic/Study 1)
2. some patients with massive PE don't even have hypoxia (Pic/Study 2)
ncbi.nlm.nih.gov/pubmed/4266893
ncbi.nlm.nih.gov/pubmed/893777
Two clues that the mechanism isn't related to the thrombus itself (i.e., decreased Q):
1. PaO2 varies widely, even in those with large PEs (Pic/Study 1)
2. some patients with massive PE don't even have hypoxia (Pic/Study 2)
ncbi.nlm.nih.gov/pubmed/4266893
ncbi.nlm.nih.gov/pubmed/893777

6/
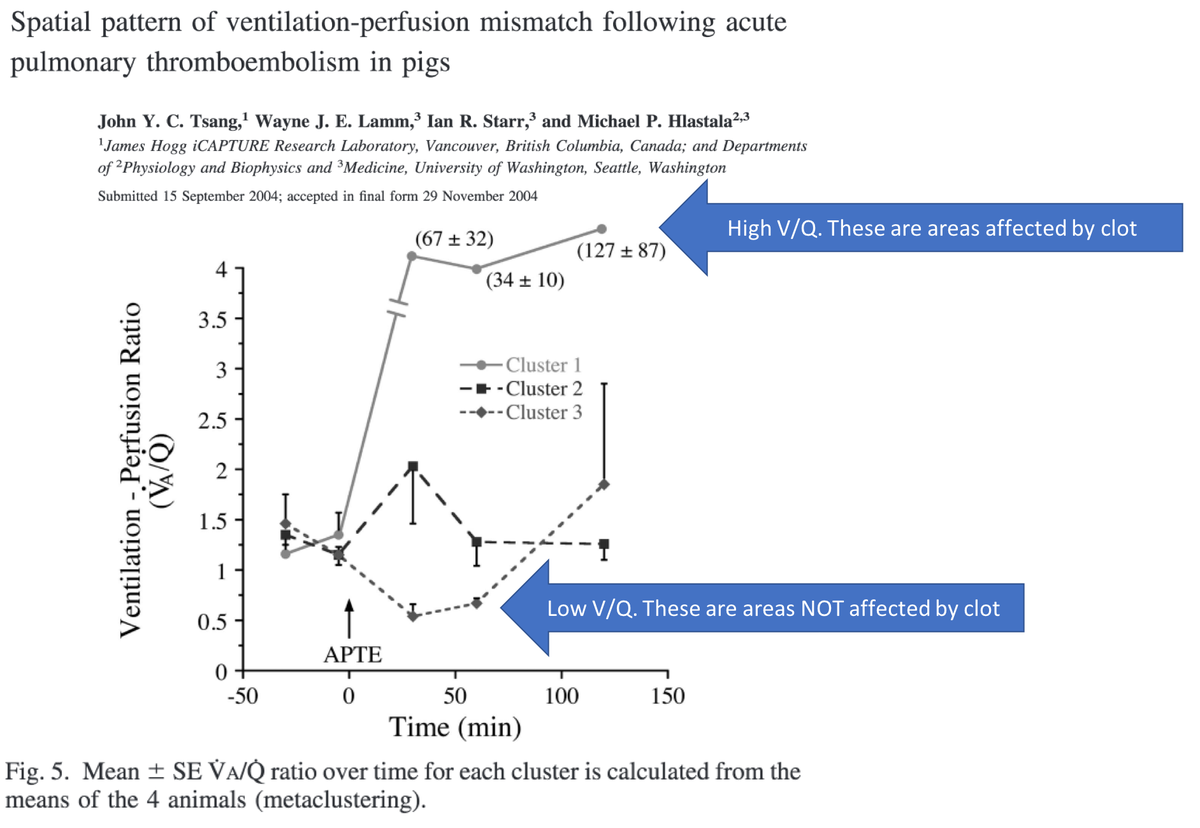
In 1984, Burton et al proposed that hypoxia resulted from V/Q mismatch in areas of the lung that were NOT affected by an embolus.
Something was going on away from where the clot had lodged.
In 1984, Burton et al proposed that hypoxia resulted from V/Q mismatch in areas of the lung that were NOT affected by an embolus.
Something was going on away from where the clot had lodged.

7/
Something is happening away from the clot. In acute PE, there are two key areas of the lung:
1. High V/Q (i.e., deadspace). These are the areas affected by the clot.
2. Low V/Q. These are the areas NOT affected by the clot.
ncbi.nlm.nih.gov/pubmed/15591291
Something is happening away from the clot. In acute PE, there are two key areas of the lung:
1. High V/Q (i.e., deadspace). These are the areas affected by the clot.
2. Low V/Q. These are the areas NOT affected by the clot.
ncbi.nlm.nih.gov/pubmed/15591291
8/
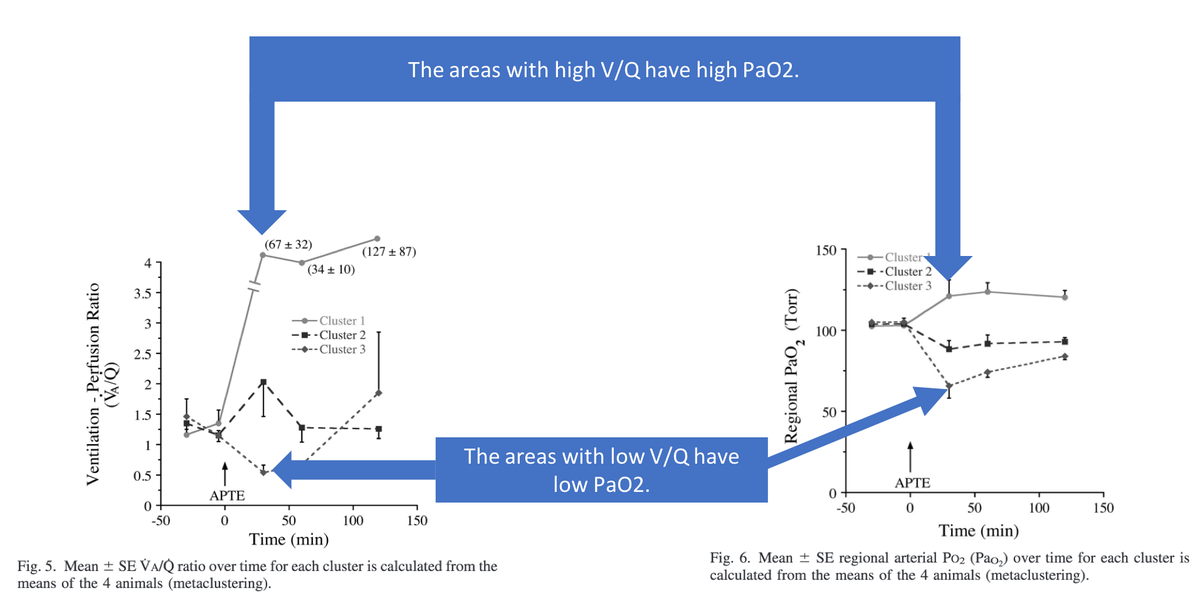
Unsurprisingly, the areas of high V/Q have high regional PaO2. In fact, the PaO2 increases after the clot lodges!
And the areas of low V/Q (again, areas not affected by clot) have low regional PaO2. These are the areas that contribute to hypoxia in acute PE.
Unsurprisingly, the areas of high V/Q have high regional PaO2. In fact, the PaO2 increases after the clot lodges!
And the areas of low V/Q (again, areas not affected by clot) have low regional PaO2. These are the areas that contribute to hypoxia in acute PE.
9/
What causes this? In short, the blood that is "blocked" by the embolus is diverted to other areas of the lung. This:
*increases Q to those areas, with
*no matching change in V to those areas
Result: ⬇️V/Q ratio (by virtue of ⬆️Q)!
And: hypoxia!
ncbi.nlm.nih.gov/pubmed/9843561
What causes this? In short, the blood that is "blocked" by the embolus is diverted to other areas of the lung. This:
*increases Q to those areas, with
*no matching change in V to those areas
Result: ⬇️V/Q ratio (by virtue of ⬆️Q)!
And: hypoxia!
ncbi.nlm.nih.gov/pubmed/9843561

10/
Using this explanation, what would you predict would happen if one used dobutamine to increased cardiac output in someone with acute PE?
Using this explanation, what would you predict would happen if one used dobutamine to increased cardiac output in someone with acute PE?
11/
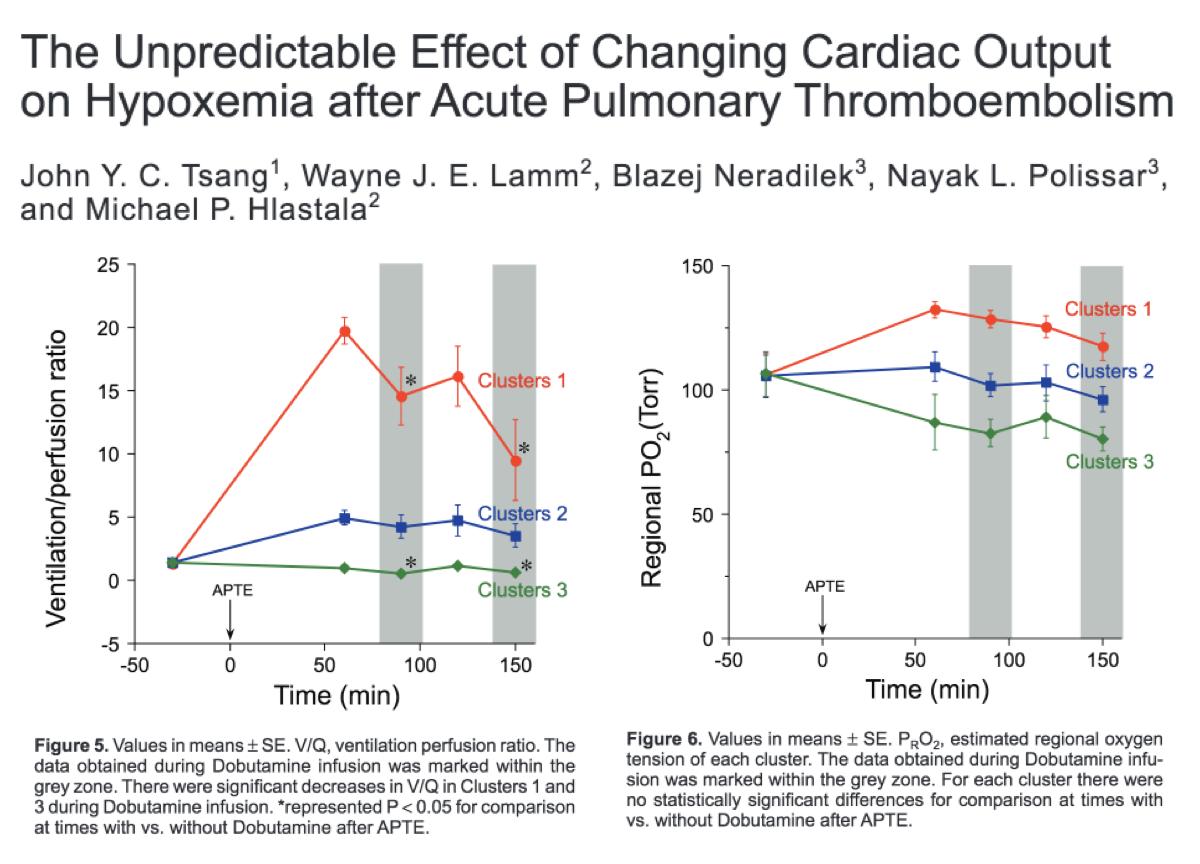
At least one experiment suggests that V/Q decreases with an increase in cardiac output! The increase in Q was not offset by an increase in ventilation.
There was no consistent effect on PaO2 in this study, despite the decreased V/Q.
journals.sagepub.com/doi/abs/10.413…
At least one experiment suggests that V/Q decreases with an increase in cardiac output! The increase in Q was not offset by an increase in ventilation.
There was no consistent effect on PaO2 in this study, despite the decreased V/Q.
journals.sagepub.com/doi/abs/10.413…
12/
The "diverted blood" theory of hypoxemia might also explain why some patients with massive PE and shock don't have hypoxia (see tweet 5).
If the blood hasn't diverted, it:
*doesn't contribute to systemic perfusion ➡ shock
*doesn't cause increased Q ➡ normal PaO2
The "diverted blood" theory of hypoxemia might also explain why some patients with massive PE and shock don't have hypoxia (see tweet 5).
If the blood hasn't diverted, it:
*doesn't contribute to systemic perfusion ➡ shock
*doesn't cause increased Q ➡ normal PaO2
13/
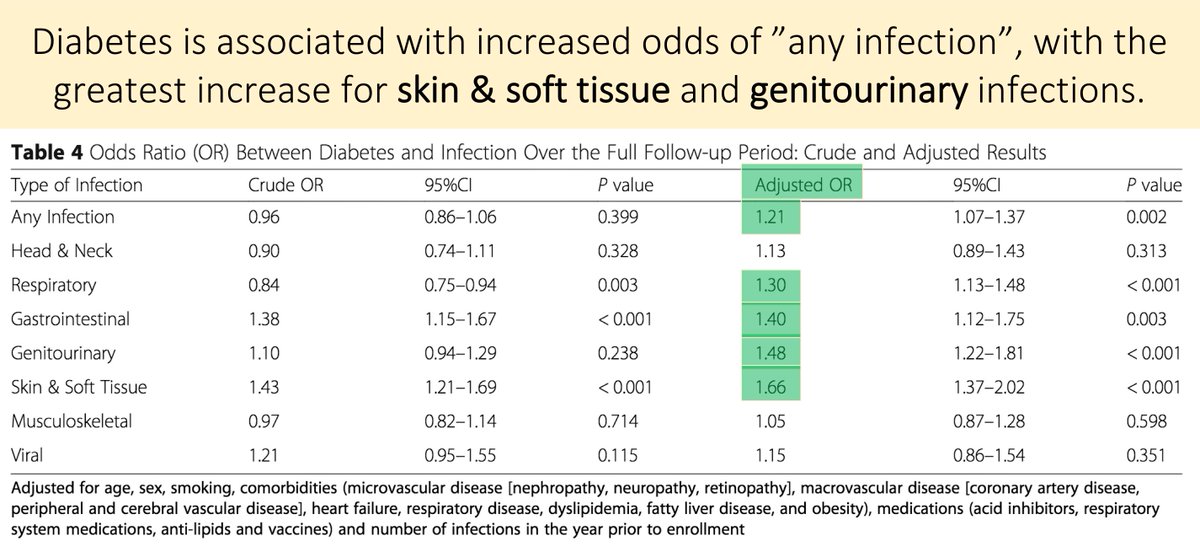
There are certainly other possible mechanisms for hypoxia in acute PE. Things like:
*atelectasis
*pulmonary edema
*bronchospasm
*shunt (e.g., via a PFO)
These may contribute, but don't seem to be the primary cause.
There are certainly other possible mechanisms for hypoxia in acute PE. Things like:
*atelectasis
*pulmonary edema
*bronchospasm
*shunt (e.g., via a PFO)
These may contribute, but don't seem to be the primary cause.
14/
Let's return to one of the questions posed above.
Acute PE leads to hypoxia primarily via V/Q mismatch. But, what's altered? And, in what direction?
[V=ventilation; Q=perfusion]
Let's return to one of the questions posed above.
Acute PE leads to hypoxia primarily via V/Q mismatch. But, what's altered? And, in what direction?
[V=ventilation; Q=perfusion]
15/
Summary. In acute PE,
*blood that would otherwise perfuse the area of clot is diverted;
*this creates an increase in Q without a matching increase in V;
*this leads to a decreased V/Q ratio;
*resulting in hypoxia
Summary. In acute PE,
*blood that would otherwise perfuse the area of clot is diverted;
*this creates an increase in Q without a matching increase in V;
*this leads to a decreased V/Q ratio;
*resulting in hypoxia
16/
If you found this thread interesting/useful, here is a link to others that attempt to answer pathophysiology mysteries/questions.
If you found this thread interesting/useful, here is a link to others that attempt to answer pathophysiology mysteries/questions.
17/UPDATE
Thanks to @DavidJuurlink for pointing out an issue in the first tweet. The blood upstream of the clot hasn't yet been oxygenated (it's in the pulmonary arteries!).
But, it's "blocked" by clot. So, the question remains: how would it contribute to hypoxia?
Thanks to @DavidJuurlink for pointing out an issue in the first tweet. The blood upstream of the clot hasn't yet been oxygenated (it's in the pulmonary arteries!).
But, it's "blocked" by clot. So, the question remains: how would it contribute to hypoxia?
18/UPDATE
In order for this blood to contribute to hypoxia, it could be shunted to the arterial circulation. This has been suggested by some on this thread (eg @strain_rate).
The studies I cite above support the major mechanism being diversion of blood, leading to increase in Q.
In order for this blood to contribute to hypoxia, it could be shunted to the arterial circulation. This has been suggested by some on this thread (eg @strain_rate).
The studies I cite above support the major mechanism being diversion of blood, leading to increase in Q.