,
23 tweets,
7 min read
Read on Twitter
Our paper on off-target toxicity of anti-cancer drugs generated a lot of really great discussion! I wanted to answer some of the questions that people were asking in one place, and highlight some areas where I think that there’s more to learn.
1) Does your work mean that all RNAi experiments are crap? No, not at all. We re-analyzed whole-genome shRNA screening data that had been collected by other investigators, and these screens correctly identified HDAC6, PIM1, etc. as non-essential. 

These huge screens typically use 5+ constructs to target each gene. That does a reasonable job of controlling for false-positives (though you get more false-negatives too). I think that the real problem is when researchers use one or two constructs and call it a day.

There are also important differences between si and shRNA, and between different shRNA vectors. To put it succinctly: I think that the bar for considering a gene essential based on RNAi experiments has historically been too low, and orthogonal validation is key.
If you’re interested in the toxicity of RNAi constructs – these are some really interesting papers that carefully characterized the mechanism of a false-positive result from prior RNAi work: elifesciences.org/articles/29702 elifesciences.org/articles/38621.
2) What about all of the problems with CRISPR? Incomplete knockouts, alternative splicing, etc? – Yes, we were definitely concerned about this. To try to deal with this, we used multiple CRISPR methodologies:
We did dropout competitions, we made and analyzed clonal knockouts, and we used CRISPR-interference in several cancer types as well. All technologies have certain limitations, but by applying multiple independent approaches we can increase our confidence in our findings.
We were particularly concerned about incomplete-knockout cell lines. (E.g., ncbi.nlm.nih.gov/pubmed/30850387). For every knockout clone in this paper, the cells were transduced with two independent gRNAs targeting functional domains.
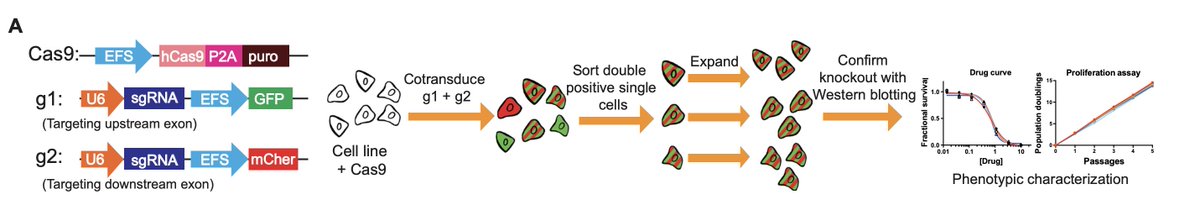
By introducing multiple mutations into a gene, we minimize the possibility that alternative splicing or downstream initiation create functional proteins. We also wrote an entire methods paper on how we make KO clones: ncbi.nlm.nih.gov/pubmed/31503414.
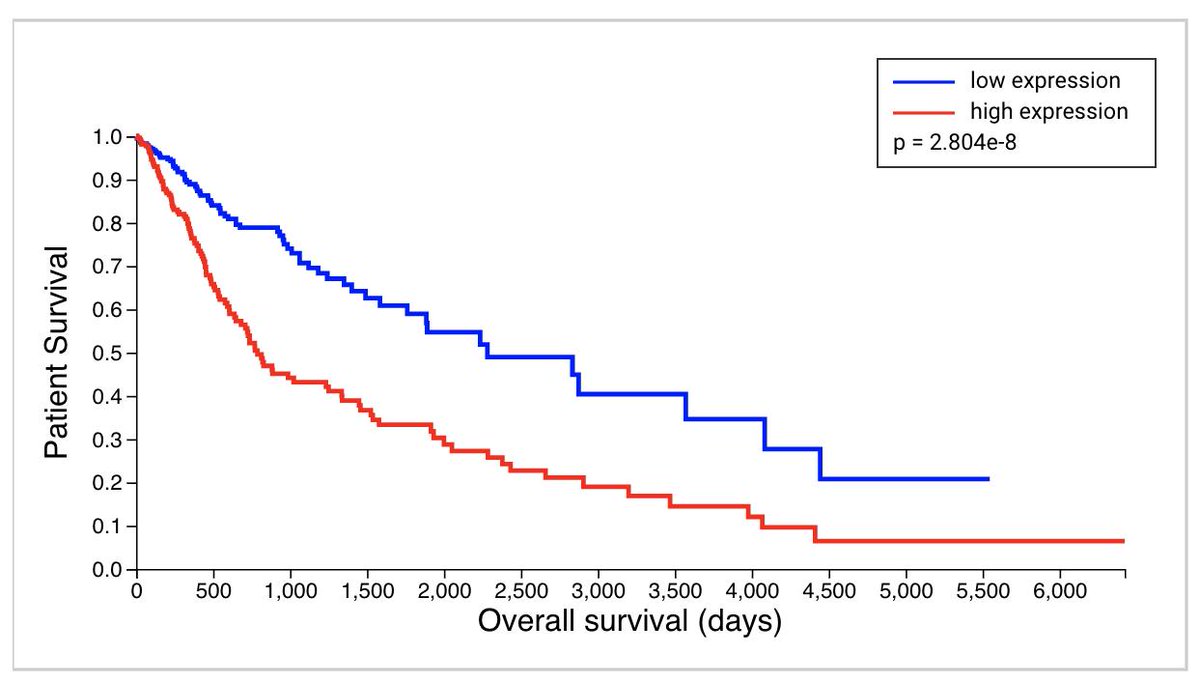
Relatedly, we show that our single-gene analysis is fully consistent with the whole-genome screening done by @JohnDoench, @TraverHart, and others:
I’d be interested in knowing – if CRISPR is prone to induce transcriptional misregulation that suppresses the effect of the mutation, is there evidence of false-negatives in these screens?
3) Does CRISPRing a gene caused its homologs to be up-regulated? (E.g., ncbi.nlm.nih.gov/pubmed/30944477). So far as I can tell, not in cancer cells. We looked in 33 KO clones that we made and didn’t see anything at all like what had been reported:

I’d be very interested to learn what other people working in cancer have observed when looking at homolog expression in CRISPR-KO’s.
4) Could these drugs be working by inhibiting their putative target’s homologs? Yes – I think that that could be the case for some of these drugs.
For the HDAC6 inhibitors – I’d bet that they are semi-specific for HDAC6 at low doses. But, cancer cells don’t care if HDAC6 is inhibited. At micromolar doses, these drugs probably inhibit other HDACs, and it’s only at those concentrations that you see anti-cancer activity.

5) Are all drugs fundamentally non-specific? It’s a tricky question, and different fields use words like “non-specific” to mean very different things.
Without forcing everyone to choose a single definition of specificity, I think that one can agree that a gradient of “specificity” exists, and some drugs are more “specific” (whatever that means) than others.
To give an example: the AstraZeneca drug AZ3146 is believed to be an inhibitor of Mps1. If we hit Mps1 with CRISPR, we get an abbreviated mitosis and massive chromosomal instability resulting in cell death.
If we treat cells with AZ3146, we see the same phenotype:


If we introduce a single point mutation into the Mps1 kinase domain, we rescue chromosome segregation and cell viability in the presence of an otherwise lethal dose of AZ3146.


AZ3146 probably binds to dozens of other cellular targets – but these experiments suggest that the major cellular effects of AZ3146 are due to its inhibition of Mps1, which can be blocked with kinase-domain mutations.
On the other hand, you have OTS167, the putative MELK inhibitor, which continues to kill cells regardless of whether MELK is expressed or not.

So, rather than totally discarding the notion of specificity, I’d suggest acknowledging that a gradient exists, and the characterization of resistance-granting mutations is good (but still far-from-perfect) evidence for in cellulo specificity.