,
17 tweets,
6 min read
Read on Twitter
1/
What causes the increase in pCO₂ in patients with acute COPD exacerbation treated with supplemental oxygen?
We routinely aim for 88-92%, fearing that higher a pO₂ values might lead to CO₂ retention. Why?
Let's look at the mechanism(s) for this feared effect.
What causes the increase in pCO₂ in patients with acute COPD exacerbation treated with supplemental oxygen?
We routinely aim for 88-92%, fearing that higher a pO₂ values might lead to CO₂ retention. Why?
Let's look at the mechanism(s) for this feared effect.
2/
In 1949, Davies reported adverse neurologic events in patients with cor pulmonale receiving O₂.
A 1955 study then demonstrated that the administration of O₂ led to an increase in CO₂. The exact mechanism was not clear.
ncbi.nlm.nih.gov/pubmed/15393115
ncbi.nlm.nih.gov/pubmed/14395425
In 1949, Davies reported adverse neurologic events in patients with cor pulmonale receiving O₂.
A 1955 study then demonstrated that the administration of O₂ led to an increase in CO₂. The exact mechanism was not clear.
ncbi.nlm.nih.gov/pubmed/15393115
ncbi.nlm.nih.gov/pubmed/14395425

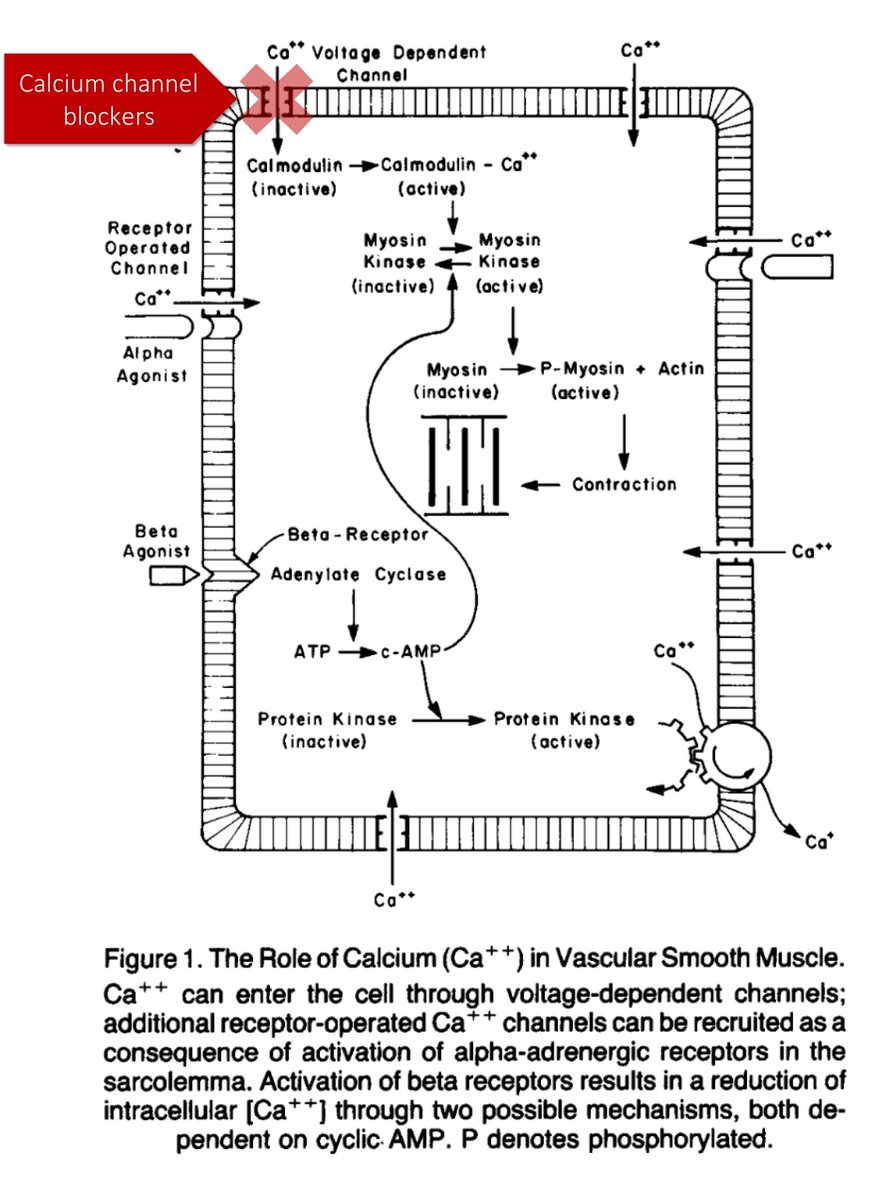
3/
What is your understanding of the predominant mechanism by which supplemental O₂ leads to increased pCO₂ in patients with acute exacerbations of COPD?
[VC = vasoconstriction]
What is your understanding of the predominant mechanism by which supplemental O₂ leads to increased pCO₂ in patients with acute exacerbations of COPD?
[VC = vasoconstriction]
4/
We've known since at least 1938 that the administration of O₂ can lead to a decrease in ventilation in patients with acute COPD.
For decades, this "loss of hypoxic drive" was felt to be the key driver of hypercarbia in these patients.
annals.org/aim/article-ab…
We've known since at least 1938 that the administration of O₂ can lead to a decrease in ventilation in patients with acute COPD.
For decades, this "loss of hypoxic drive" was felt to be the key driver of hypercarbia in these patients.
annals.org/aim/article-ab…
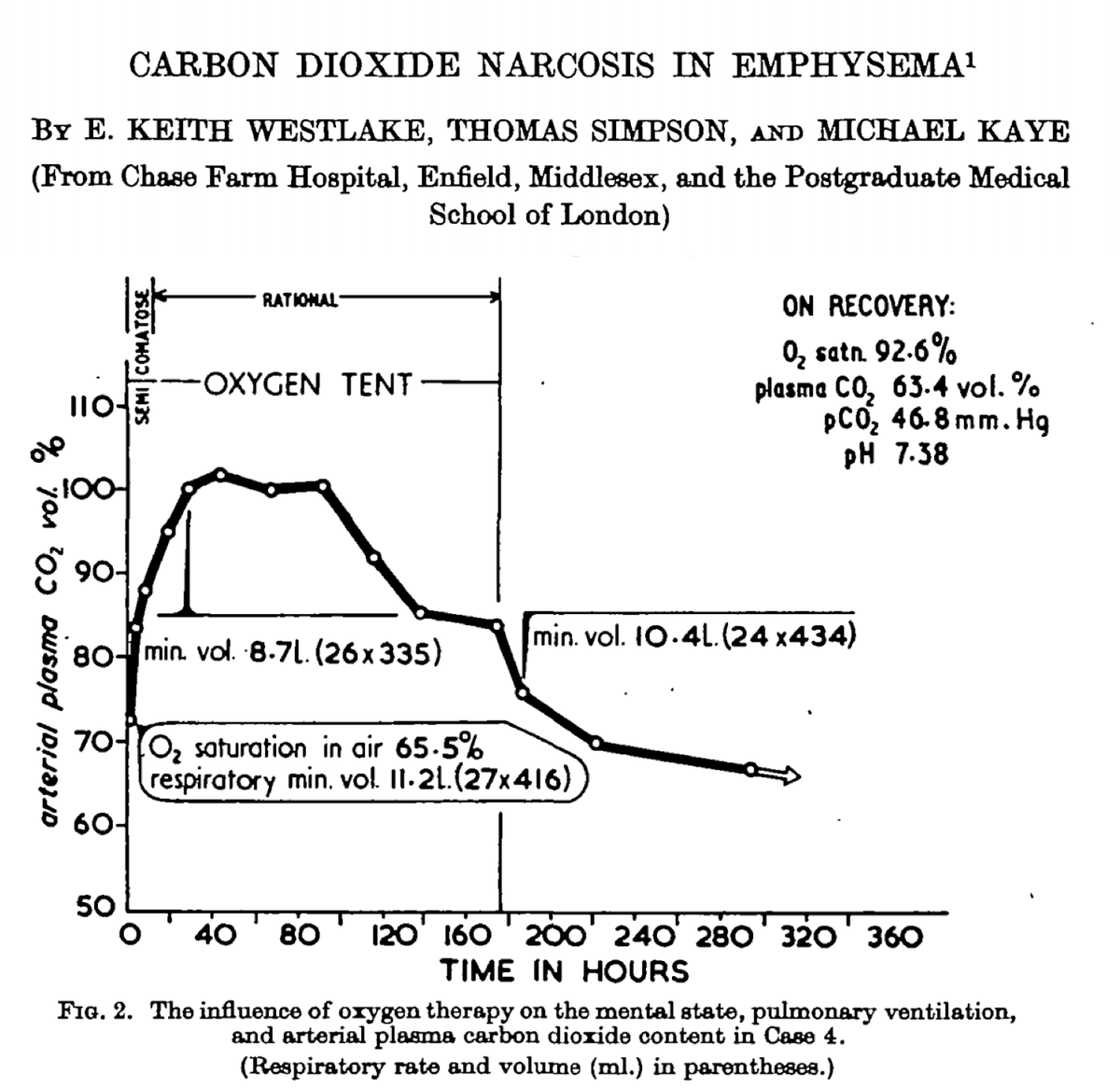
5/
In 1980, the predominance of the "loss of hypoxic drive" theory was challenged by Aubier et al. This group studied 22 patients with COPD and acute respiratory failure.
Baseline values:
pO₂ = 38 mmHg (SaO₂ ~72%)
pCO₂ = 65 mmHg
ncbi.nlm.nih.gov/pubmed/6778278
In 1980, the predominance of the "loss of hypoxic drive" theory was challenged by Aubier et al. This group studied 22 patients with COPD and acute respiratory failure.
Baseline values:
pO₂ = 38 mmHg (SaO₂ ~72%)
pCO₂ = 65 mmHg
ncbi.nlm.nih.gov/pubmed/6778278
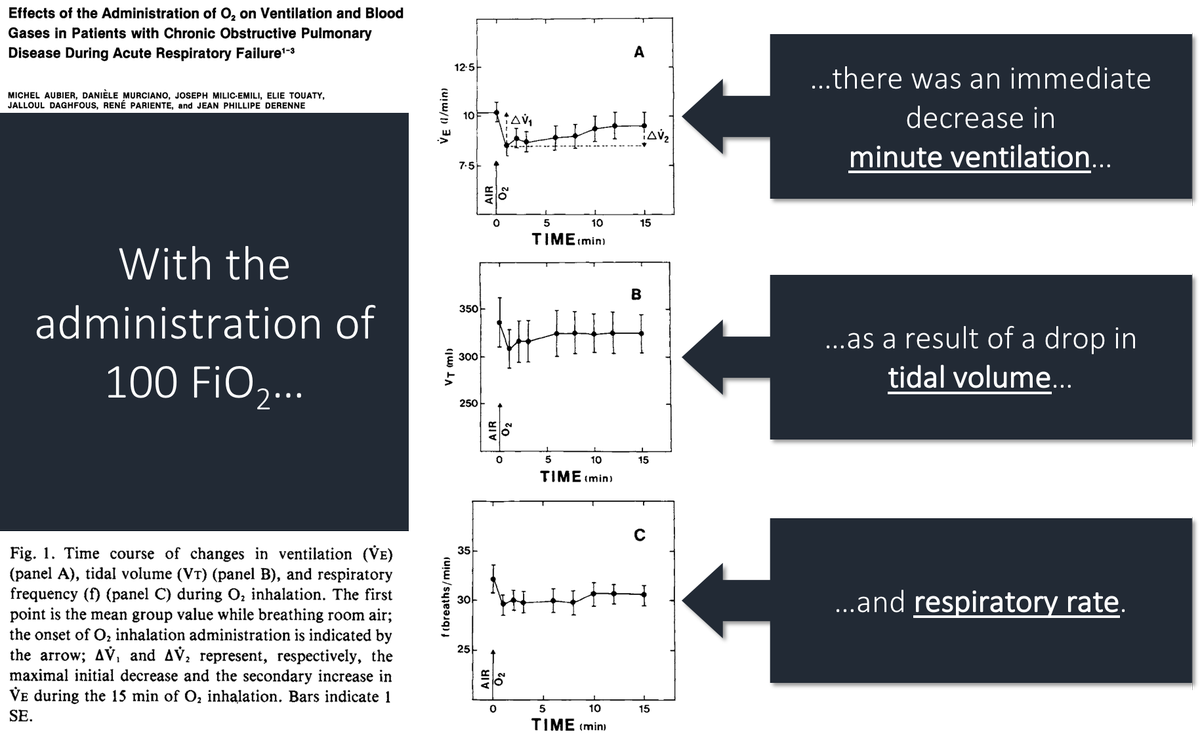
6/
After administering 100% FiO₂, they noted the following:
*pCO₂ ⬆️ by 23 mmHg
*immediate ⬇️ in ventilation by 18%
ncbi.nlm.nih.gov/pubmed/6778278
After administering 100% FiO₂, they noted the following:
*pCO₂ ⬆️ by 23 mmHg
*immediate ⬇️ in ventilation by 18%
ncbi.nlm.nih.gov/pubmed/6778278
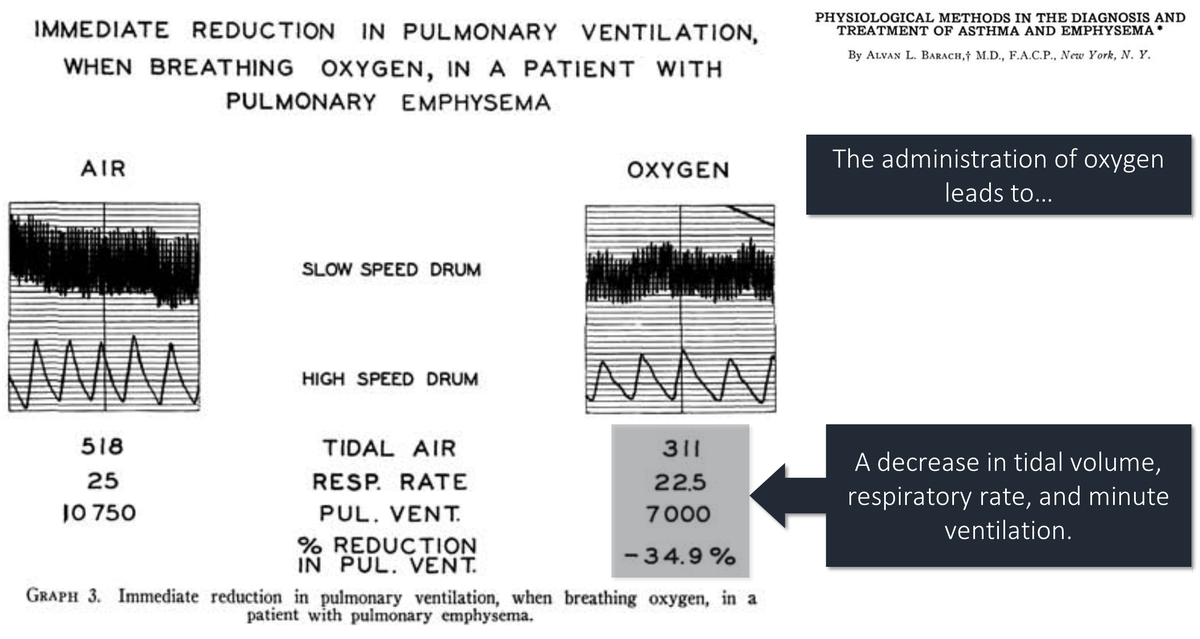
7/
But, you'll notice that ventilation returned to 93% of baseline by 15 minutes. The authors conclude that the increase in pCO₂ can be attributed to:
48% (11mmHg) = loss of hypoxic vasoconstriction
30% (7mmHg) = Haldane effect
22% (5mmHg) = loss of hypoxic drive
But, you'll notice that ventilation returned to 93% of baseline by 15 minutes. The authors conclude that the increase in pCO₂ can be attributed to:
48% (11mmHg) = loss of hypoxic vasoconstriction
30% (7mmHg) = Haldane effect
22% (5mmHg) = loss of hypoxic drive
8/
Why would the loss of hypoxic vasoconstriction lead to an increase in pCO₂?
Why would the loss of hypoxic vasoconstriction lead to an increase in pCO₂?
9/
With the loss of hypoxic vasoconstriction, blood flow (Q) is redirected away from areas of high ventilation (V). The fall in Q leads to:
⬆️ regional V/Q
⬆️ deadspace
Consistent with this, Aubier found that deadspace ventilation ⬆️ from 77% to 82% after administration of O₂.
With the loss of hypoxic vasoconstriction, blood flow (Q) is redirected away from areas of high ventilation (V). The fall in Q leads to:
⬆️ regional V/Q
⬆️ deadspace
Consistent with this, Aubier found that deadspace ventilation ⬆️ from 77% to 82% after administration of O₂.

10/
Some subsequent studies (e.g., PMID 9295826, PMID 8565533) have supported the theory that loss of hypoxic vasoconstriction and altered V/Q is the main driver of hypercarbia with the administration of O₂.
But, not all agree (e.g., PMID 3590052).
Some subsequent studies (e.g., PMID 9295826, PMID 8565533) have supported the theory that loss of hypoxic vasoconstriction and altered V/Q is the main driver of hypercarbia with the administration of O₂.
But, not all agree (e.g., PMID 3590052).
11/
This thread has addressed patients with acute COPD. Does the same apply to patients with stable COPD?
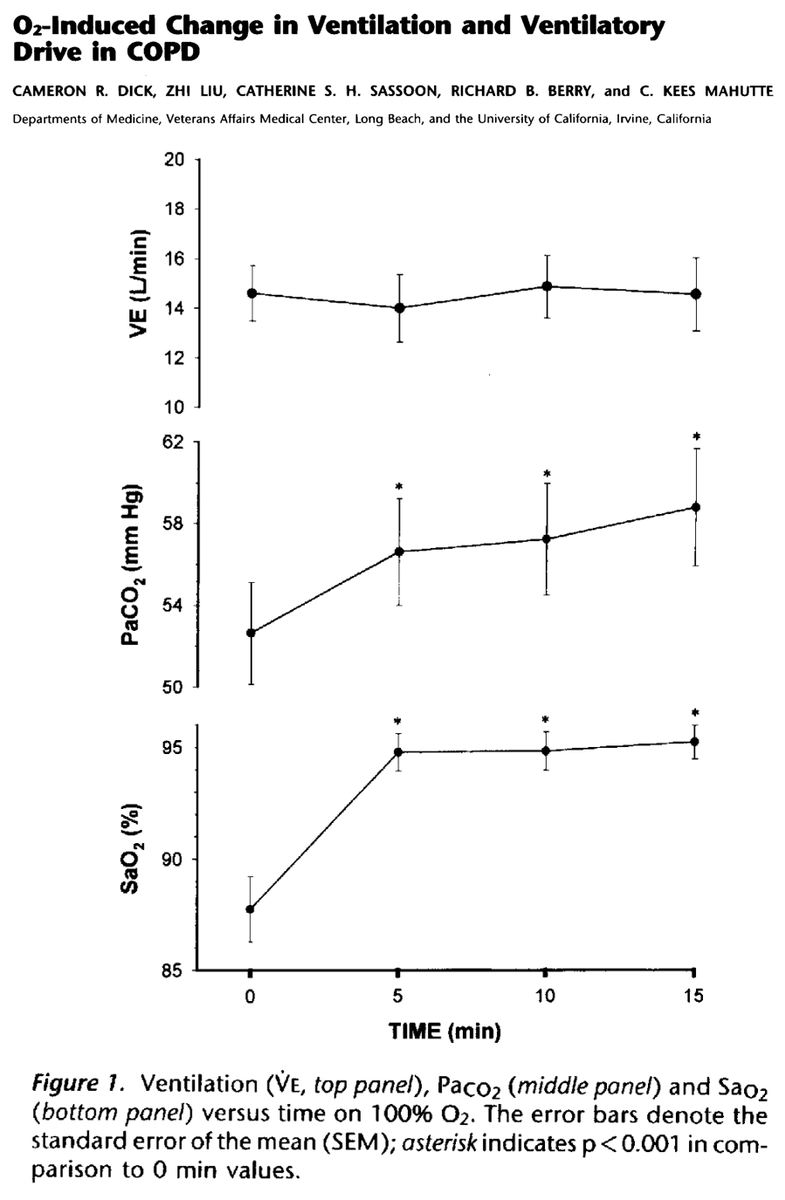
One study examined stable COPD patients breathing 100% O₂ for 15min:
Ventilation ⬇️ by 0.08 L/min (this is tiny)
pCO₂ ⬆️ by 6.6mmHg (also small)
ncbi.nlm.nih.gov/pubmed/9032202
This thread has addressed patients with acute COPD. Does the same apply to patients with stable COPD?
One study examined stable COPD patients breathing 100% O₂ for 15min:
Ventilation ⬇️ by 0.08 L/min (this is tiny)
pCO₂ ⬆️ by 6.6mmHg (also small)
ncbi.nlm.nih.gov/pubmed/9032202
12/
What explains the small change in ventilation and pCO₂ seen in stable COPD patients?
One reason: the decrease in ventilation one might expect from the administration of 100% O₂ was "offset" by the appropriate increase in ventilation in response to an increase in pCO₂
What explains the small change in ventilation and pCO₂ seen in stable COPD patients?
One reason: the decrease in ventilation one might expect from the administration of 100% O₂ was "offset" by the appropriate increase in ventilation in response to an increase in pCO₂

13/
Compare this with patients with acute COPD.
They are already breathing at or near their maximal sustainable ventilation. They are therefore are unable to augment their ventilation in response to an increase in pCO₂.
Compare this with patients with acute COPD.
They are already breathing at or near their maximal sustainable ventilation. They are therefore are unable to augment their ventilation in response to an increase in pCO₂.
14/
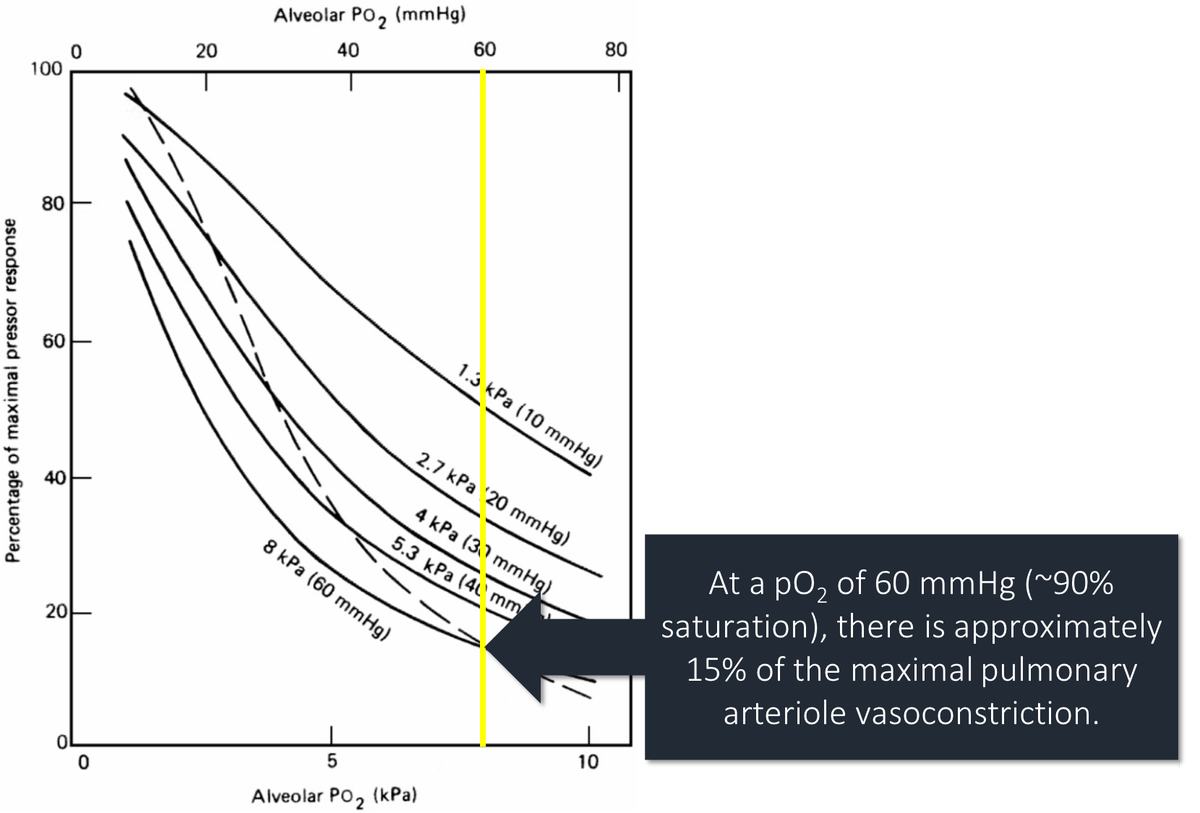
Another potential difference is the degree of hypoxia.
At a pO₂ of 60mmHg vasoconstriction is 15% of the max. As pO₂ falls, vasoconstriction increases rapidly. Stable patients, with less severe hypoxia, may not have much vasoconstriction to "lose".
ncbi.nlm.nih.gov/pubmed/9295826
Another potential difference is the degree of hypoxia.
At a pO₂ of 60mmHg vasoconstriction is 15% of the max. As pO₂ falls, vasoconstriction increases rapidly. Stable patients, with less severe hypoxia, may not have much vasoconstriction to "lose".
ncbi.nlm.nih.gov/pubmed/9295826
15/
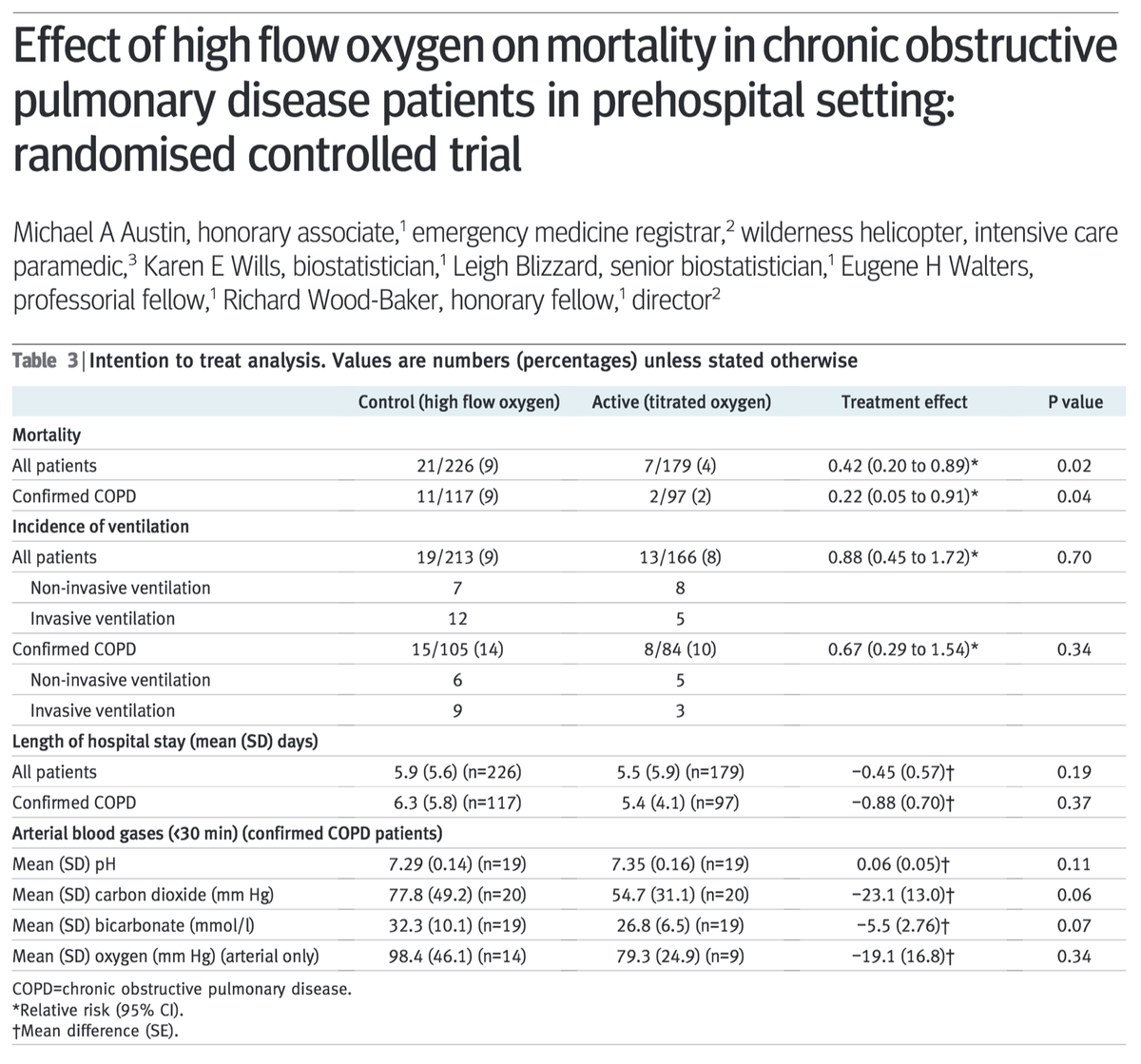
Regarding our goal SaO₂ of 88-92%, this partly comes from RCT data comparing O₂ titrated to this range versus high flow O₂. One study of 405 patients with acute COPD reported mortality of:
9% for high flow O₂
4% for titrated O₂
HR 0.48 (p=0.02)
ncbi.nlm.nih.gov/pubmed/20959284
Regarding our goal SaO₂ of 88-92%, this partly comes from RCT data comparing O₂ titrated to this range versus high flow O₂. One study of 405 patients with acute COPD reported mortality of:
9% for high flow O₂
4% for titrated O₂
HR 0.48 (p=0.02)
ncbi.nlm.nih.gov/pubmed/20959284
16/
Before closing, let's ask the original question again (note: there may not be a "right" answer).
What is your understanding of the predominant mechanism by which supplemental O₂ leads to increased pCO₂ in patients with acute exacerbations of COPD?
[VC = vasoconstriction]
Before closing, let's ask the original question again (note: there may not be a "right" answer).
What is your understanding of the predominant mechanism by which supplemental O₂ leads to increased pCO₂ in patients with acute exacerbations of COPD?
[VC = vasoconstriction]
17/
Summary:
⭐️hypercarbia after O₂ administration in acute COPD is multifactorial
⭐️loss of "hypoxic drive" may be a minor contributor
⭐️loss of hypoxic vasoconstriction is likely key
⭐️the above may be less applicable in those with stable COPD
⭐️RCT data supports titrated O₂
Summary:
⭐️hypercarbia after O₂ administration in acute COPD is multifactorial
⭐️loss of "hypoxic drive" may be a minor contributor
⭐️loss of hypoxic vasoconstriction is likely key
⭐️the above may be less applicable in those with stable COPD
⭐️RCT data supports titrated O₂